1Fondazione Smith Kline, Verona
2Dipartimento di Informatica – Università di Pisa COSBI – Rovereto, Italy. Vydiant – CA, USA
3Istituto di Management – Scuola Superiore Sant’Anna (Pisa)
4Healthcare & Life Science, IBM Italia
5Medicine Science and Technology, GlaxoSmithKline, Stevenage (UK)
Tendenze Nuove n.1 – 2019; 1-21: DOI: 10.32032/TENDENZE201908.PDF
 Scarica Pdf
Scarica Pdf
Abstract
Background: il corretto riconoscimento del bersaglio biologico, del sistema patologico e delle caratteristiche del composto sono requisiti indispensabili per uno sviluppo del farmaco ragionato e quindi pianificato sulle conoscenze a priori piuttosto che basato su evidenze empiriche osservazionali. Il numero di esperienze scientifiche che documentano la capacità delle tecnologie computazionali di gestire i dati della ricerca farmaceutica al fine di accelerare ed ottimizzare ogni fase dello sviluppo di un nuovo farmaco è in crescente aumento.
Obiettivo: rassegna della letteratura sul ruolo dell’Intelligenza Artificiale, e delle metodiche appartenenti quali il machine learning e deep learning, nei processi dello sviluppo preclinico del farmaco.
Risultati: queste tecnologie risultano largamente in uso nella ricerca di nuove terapie sia in contesto pubblico che privato e si sono ritagliate un ruolo significativo nei processi di drug design de novo, virtual screening, analisi della relazione quantitativa struttura-attività (QSAR), valutazione in silico del profilo di assorbimento, distribuzione, metabolismo, escrezione e tossicità (ADME/T). Recentemente le metodiche di deep learning, suffragate dalle prestazioni superiori rispetto ad altri algoritmi di apprendimento automatico, stanno mostrando risultati promettenti nell’affrontare e superare diverse sfide nella scoperta di nuovi farmaci.
Limiti: le prove di evidenza sono di difficile confronto in quanto differiscono per algoritmi, fonte dei dati e metodi di training.
Conclusioni: le evidenze documentate sono di significativo interesse ed alimentano l’importante aspettativa di mettere a disposizione dei pazienti nuovi composti più accessibili, in minor tempo e sostanziati da dati che ne permettano una più efficiente trasferibilità alla fase di sviluppo clinico e quindi alla real life clinica.
Parole chiave: artificial intelligence; machine learning; drug discovery; chemoinformatics; de novo design; deep learning; neural networks; property prediction; quantitative structure-activity relationship (QSAR);
Introduzione
La ricerca & sviluppo (R&S) dei farmaci è un processo costoso, lungo ed inefficiente, che richiede fino a 15 anni per trasferire al mercato un nuovo farmaco, con un costo medio di 2.5 miliardi di dollari statunitensi ed un rischio di fallimento elevato (1). In media solo il 13.8% dei composti che entrano nella fase 1 di sviluppo clinico arriva all’approvazione, con tassi di successo minimi del 3.4% in campi di ricerca ad alto bisogno terapeutico come nel caso dei farmaci oncologici (2).
La necessità di superare i limiti nello sviluppo di nuove terapie è resa ancor più evidente alla luce dei dati di fabbisogno di salute globale, con la fascia di popolazione mondiale over 60 stimata a raggiungere il 25% della popolazione entro il 2050 e della carenza di molecole approvate per il 95% delle oltre 7000 malattie rare conosciute (3,4).
Metà dei fallimenti sono dovuti a mancanza di efficacia, mentre ben un quarto dei fallimenti sono dovuti a problematiche di tollerabilità, entrambe le cause ad espressione della difficoltà di selezionare il giusto target per la malattia in studio (5).
Al fine di ottimizzare il processo di scoperta e sviluppo di nuove molecole terapeutiche è dunque necessario utilizzare nella maniera più efficiente la conoscenza nascosta nella complessità dei dati messi a disposizione dalla ricerca biomedica. Tali dati tuttavia sono eterogenei, derivano da fonti tra loro diverse, pubbliche o private, quali sequenziamento genomico, proteomica, database sanitari, social networks, wearable devices, database bibliografici come Medline e banche di composti chimico-farmaceutici.
La gestione di tale mole di informazioni, in parte ancora in forma di dati non-strutturati, definiti nel complesso Big Data a motivo della dimensione e complessità, rappresenta in ogni ambito disciplinare una sfida, prima ancora della loro analisi ed utilizzo.
Grazie alle capacità computazionali e delle metodologie dell’informatica e delle tecniche di apprendimento automatico è però possibile gestire ed analizzare in maniera automatizzata il volume di dati biomedici, al fine di estrapolarne relazioni significative, generare nuove ipotesi da sottoporre a verifica sperimentale e prevedere con metodo statistico l’accadimento di fenomeni futuri, compresi efficacia e tossicità associati ai farmaci.
Nel campo della ricerca farmaceutica l’Intelligenza Artificiale (Artificial Intelligence, AI) applica algoritmi computazionali ai fini di analisi, apprendimento ed esplicazione dei big data farmaceutici utili ad individuare nuove molecole (6).
L’applicazione di metodi computazionali alla ricerca farmaceutica non è una novità di questi ultimi anni: nell’ottobre 1981 la rivista Fortune pubblica in copertina un articolo intitolato “Next Industrial Revolution: Designing Drugs by Computer at Merck”, evento considerato da diversi autori come l’inizio dell’interesse per le potenzialità dello sviluppo razionale di nuove molecole con l’ausilio di strumenti computerizzati (7).
Da allora il palesarsi dei limiti dell’high-throughput screening (HTS), che nasceva e si sviluppava proprio negli stessi anni, in associazione ad una drastica riduzione dei costi di stoccaggio e gestione dei dati, ha rinnovato interesse, applicazioni ed aspettative di metodiche di chemio-bioinformatica basate su algoritmi automatizzati.
La presente revisione della letteratura ha lo scopo di introdurre i principi e fondamenti di AI, le applicazioni alla scoperta e sviluppo di molecole candidate a divenire nuove terapie, con un focus specifico sulle applicazioni delle metodiche di Deep Learning (DL). Nello specifico verranno esposte le attuali evidenze relative alla ricerca e sviluppo preclinico di nuove molecole, quali i processi di target identi cation, Hit discovery, Hit-to-lead optimization, analisi delle proprietà ADME/T ed identificazione dei composti candidati alla sperimentazione clinica.
Artificial Intelligence, Machine Learning, Deep Learning
L’esplosione di dati e la loro disponibilità in continuo aggiornamento (8), in associazione al miglioramento delle capacità di calcolo delle tecnologie informatiche, ha favorito il rifiorire di tecniche utili all’estrazione di informazioni significative da grandi quantità di dati attraverso metodi automatici o semi-automatici, processo che prende il nome di Data Mining (DM).
Un concetto correlato al data mining è quello di apprendimento automatico o Machine Learning (ML), processo tramite il quale i computer possono, utilizzando algoritmi interattivi, imparare dai dati, descrivere e predire risultati senza che essi siano stati specificamente programmati per questo fine grazie al riconoscimento automatico di schemi (patterns) tra i dati.
Sebbene sia il DM che il ML utilizzino entrambi modelli statistici per comprendere le caratteristiche delle variabili e derivarne inferenze, gli strumenti di DM hanno l’obiettivo di descrivere, esplicare e aiutare l’operatore nella comprensione dei dati, mentre il ML, che afferisce all’area dell’Artificial Intelligence, permette non solo di elaborare autonomamente patterns dai dati, ma anche di costruire nuovi modelli prescrittivi e predittivi.
L’apprendimento della macchina può definirsi supervisionato, non- supervisionato o rinforzato. Nel primo caso le variabili di input e output sono già etichettate e classificate e la macchina analizza i dati basandosi sulla correlazione tra patterns ed outcomes dei dati in possesso. Nel caso dell’apprendimento non supervisionato, i dati non sono classificati a causa della mole ed eterogeneità (social media ed esempio) e quindi subiscono un primo processo di identificazione di patterns, clusterizzazione e classificazione per poi essere analizzati secondo modelli supervisionati. L’apprendimento rinforzato è un modello di apprendimento esperienziale nel quale l’algoritmo della macchina non attinge da un set di dati predefinito, ma apprende sulla base del feedback ricevuto da azioni errate o di successo in base ad un determinato task. Quest’ultimo approccio è classicamente utilizzato in robotica nella locomozione autonoma e nei sistemi di guida automatizzata, ma può anche essere utilizzato in combinazione con altri tipi di apprendimento automatico.
I metodi di ML quali regressioni logistiche, Support Vector Machine (SVM), k-nearest neighbors (k-NN), Naïve Bayesian (NB) o alberi decisionali, sono stati utilizzati per anni dalla ricerca farmaceutica (9), ma è stata la recente esplosione di dati e la loro complessità a richiedere l’utilizzo di nuovi approcci quali il Deep Learning (DL).
Il termine DL definisce una specifica e più recente modalità di ML che utilizza reti neuronali artificiali (artificial neuronal networks, ANNs) a strati (layers) multipli di unità processanti non lineari al fine di comprendere i dati in modalità iterativa. La struttura elementare delle ANNs (figura 1) è basata sulla presenza di più neuroni artificiali (nodi) di input, uno o due strati di nodi nascosti che elabora i segnali degli input con funzioni di attivazioni e pesi diversi, ed uno o più nodi di output. Le ANNs moderne si ispirano al modello di architettura neuronale della corteccia encefalica umana ed il loro addestramento avviene attraverso le modifiche iterative dei pesi e delle funzioni di attivazione, al fine di minimizzare l’errore tra il risultato e l’output atteso (10).
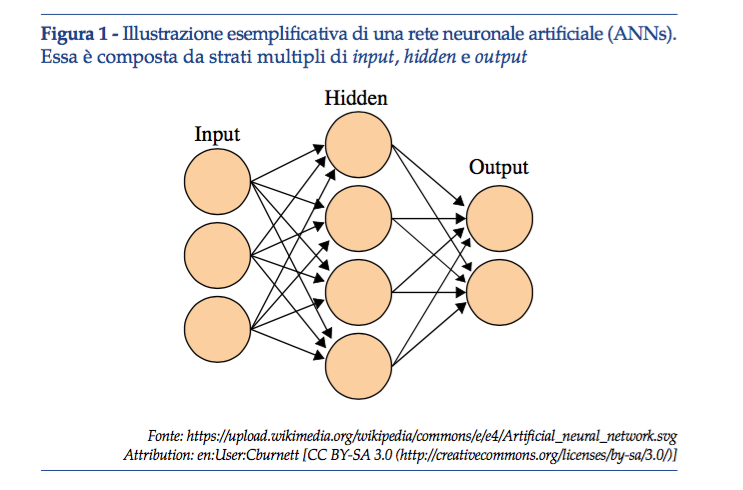
Tale modello è costituito da un gruppo di interconnessioni di neuroni artificiali detti nodi che utilizzano un approccio di connessionismo di calcolo. Nella maggior parte dei casi una rete neurale artificiale è un sistema adattivo che cambia la sua struttura basata su informazioni esterne o interne che scorrono attraverso la rete durante la fase di apprendimento. I nodi compongono i layers e sono connessi a vario grado con i neuroni degli strati adiacenti. Attraverso equazioni complesse le variabili sono acquisite dai nodi input, trasmesse ed elaborate ai nodi output attraverso i nodi hidden.
Al fine di prevenire problemi di overfitting statistico, il modello DL o Deep Neural Network (DNN) utilizza un numero di strati nascosti che variano da 5 a 1000 a seconda della tipologia dell’analisi e la principale differenza tra DL e ANN tradizionali risiede nella grandezza, complessità delle reti neuronali e di conseguenza della potenza di calcolo richiesta.
ML e DL sono categorie dell’Intelligenza Artificiale, branca dell’informatica impegnata nella progettazione di software capaci di fornire prestazioni che, a un osservatore comune, sembrerebbero essere di pertinenza esclusiva dell’intelligenza umana come reasoning (abilità di risolvere i problemi sulla base della logica deduzione), knowledge (comprensione di specifiche entità nell’ambito del contesto), planning (abilità di raggiungere un obiettivo), communication (capacità di comprendere il linguaggio scritto e verbale), perception (capacità di dedurre sulla base di input sensoriali visivi o uditivi) (11).
I modelli di DL apprendono a partire da dati non strutturati tramite algoritmi non supervisionati e trovano per questo motivo un campo di applicazione molto vasto. Ad esempio, in medicina, il DL sta ottenendo i primi risultati significativi in aree come la radiologia, l’anatomia patologica, la dermatologia, l’oftalmologia, la salute mentale, la stratificazione del rischio e la ricerca farmaceutica (12).
Nell’ultimo decennio, il DL ha ottenuto un notevole successo in varie aree di ricerca sull’intelligenza artificiale, soprattutto attraverso modelli di reti neurali convoluzionali (CNN) per il riconoscimento di immagini e reti neurali ricorrenti (RNN) per il riconoscimento di testi. Evoluta dalla precedente ricerca sulle reti neurali artificiali, questa tecnologia ha mostrato prestazioni superiori rispetto ad altri algoritmi di apprendimento automatico in aree quali il riconoscimento di immagini e voce, l’elaborazione del linguaggio naturale. La potenzialità del DL alla ricerca farmaceutica è emersa negli ultimi anni e la sua applicazione ha mostrato risultati promettenti nell’affrontare e superare diverse sfide nella scoperta di nuovi farmaci (13).
Target Identification
I modelli fisiopatologici classici basati su una consecutio lineare di eventi che descrivono cambiamenti che portano un sistema biologico fisiologico ad una condizione patologica sono ormai superati alla luce delle evidenze. La patogenesi e la patofisiologia delle principali patologie croniche rimangono ad oggi multifattoriali, complesse e parzialmente sconosciute. Il ruolo patogenetico di polimorfismi genetici, infezioni virali latenti, fattori ambientali, alterazioni dell’asse neurovegetativo e soprattutto quadri di infiammazione cronica subclinica sono trasversalmente riconosciuti nella fisiopatologia di malattie cardiovascolari, respiratorie, neurodegenerative, autoimmunitarie e in tutte le fasi di sviluppo e progressione del cancro (14). Solo composti farmaceutici capaci di agire sui fattori primordiali, multipli e complessi, dei processi patologici potranno interferire nei meccanismi profondi d’insorgenza della malattia e portare a guarigione intesa come restitutio ad integrum del sistema o apparato. Farmaci che al contrario agiscono su parte dei meccanismi o su meccanismi consequenziali ai primordiali portano ad una riduzione della sintomatologia e a una dilatazione temporale dell’insorgenza di complicanze ed esiti di malattia mantenendo quindi una condizione di cronicità. È documentato ad esempio come anche in caso di un controllo intensivo farmacologico della glicemia in pazienti diabetici si manifestino complicanze micro e macro vascolari, a testimonianza di come nella patologia del diabete mellito, al di là della documentata glucotossicità, ci siano altri fattori che agiscono prima e durante lo stato iperglicemia (15).
I processi decisionali di prioritizzazione e selezione delle indicazioni delle molecole in sviluppo dovranno dunque sempre più basarsi su informazioni significative estrapolate dall’insieme complessivo delle interazioni molecolari in una particolare cellula, definito interattoma, integrate ai medesimi dati provenienti da tessuti, organi, apparati e sistemi nel loro insieme. Il lavoro di Nelson et coll. nel 2015 stima infatti che la selezione di target biologici sostanziata da dati genetici a supporto raddoppi le possibilità di successo nella fase I dello sviluppo clinico (16). Una rianalisi del 2019 ampliata e potenziata con analisi statistiche specifiche per target conferma questi risultati e sottolinea nuovamente la necessità di investimenti in acquisizione, integrazione ed analisi di dati genomici (17).
Knowledge extraction, biomarker identification and pathway analysis
Una pipeline ideale per acquisire maggiore conoscenza dai dati disponibili prevede almeno tre passi da eseguire in sequenza (18). Il primo passo, detto knowledge extraction, serve ad integrare i dati disponibili sperimentalmente con quanto si riesca ad ottenere dal pubblico dominio mediante la scansione automatica della letteratura, dei brevetti e dei database specializzati. Questo passo prevede l’uso di tecniche di intelligenza artificiale come il natural language processing per estrarre relazioni tra le entità dei modelli che si vogliono costruire relativi alla patologia di interesse. L’integrazione di tutte le informazioni acquisite è la base di conoscenza da cui partire per individuare biomarcatori (insiemi di molecole la cui variazione quantitativa consente di effettuare diagnosi, prevedere prognosi, individuare la classe di soggetti che rispondono a una terapia, i sottotipi di una stessa patologia, ecc.) per stratificare i soggetti in classi che abbiano caratteristiche omogenee e che consentano quindi di muoverci verso terapie mirate (19). I biomarcatori sono spesso individuati ignorando ogni aspetto meccanicistico dell’evoluzione della patologia o i meccanismi di azione dei farmaci. Per comprendere meglio come mai un biomarcatore funziona occorre muoverci ad un livello di analisi più sistemico e considerare i pathway di segnalazione molecolare. Partendo da un interattoma su cui mappare i dati sperimentali disponibili e i biomarcatori individuati, tecniche di biologia delle reti e algoritmi di flusso di informazione consentono di evidenziare le sottoreti affette dalla patologia connettendo dati sperimentali e biomarcatori sui grafi. È inoltre possibile fare una graduatoria dei pathway individuati considerando quelli più attivi nelle condizioni considerate (20). Questo aspetto è essenziale nella comprensione del meccanismo di azione dei farmaci o nell’individuazione di nuovi target.
Predizione delle interazioni tra proteine
I metodi sperimentali per determinare la struttura delle proteine sono lenti e costosi, e possono essere applicati solo a una piccola porzione (<0,1%) delle proteine prodotte da vari progetti di sequenziamento genomico. Pertanto, metodi affidabili per predire la struttura delle nuove proteine scoperte usando le loro sequenze amminoacidiche sono di cruciale importanza per accelerare la ricerca per determinare il ruolo di queste proteine nei sistemi biologici. Sebbene nella maggior parte dei casi la conoscenza della sequenza amminoacidica di una proteina sia nota, la predizione accurata della struttura tridimensionale de novo di una proteina resta una sfida.
Recentemente architetture di DL sono state applicate per predire con successo la struttura secondaria proteica ab initio e sono considerate la metodica più promettente nell’ottenere predizioni accurate di strutture proteiche 3D a partire da sequenza amminoacidi nota (21).
Le interazioni proteina-proteina sono determinanti nella maggior parte dei processi patologici e rappresentano uno spazio biochimico sin ora largamente inesplorato dove possono agire nuovi composti capaci di modularne l’attività. In effetti i metodi tradizionali di screening di librerie di composti chimici hanno funzionato nel bersagliare il sito attivo proteico, ma si sono dimostrati di scarso successo nell’individuare ligandi chimici capaci di modulare le interazioni proteina-proteina. Dall’analisi dell’interfaccia di interazione proteina-proteina è possibile individuare una nuova classe di bersagli molecolari che differiscono da bersagli tradizionali come i recettori accoppiati a proteine G (GPCRs), canali ionici, chinasi e recettori nucleari (22). L’attuale ricerca sull’interattoma umano suggerisce che il numero di interazioni proteina-proteina sia compreso tra 130.000 e 650.000, e solo una piccola frazione di questi è stata individuata come bersaglio farmacologico (23).
In confronto ai bersagli tradizionali la modulazione delle interazioni proteina-proteina ha la potenzialità di ridurre gli eventi avversi come conseguenza della maggiore selettività biologica (24). Lo studio dello spazio biochimico nell’interfaccia proteina-proteina si basa sul docking molecolare, metodologia che esplora il comportamento di piccole molecole nell’interazione con i siti di legame in proteine target e predice le condizioni per la formazione di un complesso stabile. In questo ambito le metodiche di DL, rispetto ai metodi tradizionali, vantano una maggiore accuratezza dovuta alla capacità di estrarre automaticamente elementi significativi dalla sequenza amminoacidica del sito di legame (25).
A questo scopo i recenti modelli di DL si sono dimostrati capaci di analizzare automaticamente uno specifico punto di interesse della struttura proteica, segmentando il microambiente in sezioni dallo spessore di 1.25 1.25 Å (10-10 m) ed estraendo circa 80 proprietà fisico-chimiche per ciascuna sezione tra cui il tipo di gruppo funzionale atomico, idrofobicità e struttura secondaria (26).
Hit Discovery
Una volta individuato e validato il bersaglio biologico è necessario selezionare, a partire da una libreria di composti chimici, una serie di composti caratterizzati da affinità e attività biologica generica desiderata, ciascuno definito Hit Compound (27). Lo screening dell’Hit Compound a partire da migliaia di composti inseriti nelle librerie chimiche industriali o accademiche può avvenire tramite saggi fisici con l’utilizzo di tecniche robotiche come high throughput screening (HTS) o attraverso il Virtual Screening che utilizza algoritmi software per lo screening in silico. Le metodiche di virtual screening comprendono tra le altre il docking e il machine learning (7). Nei casi in cui sia disponibile la struttura tridimensionale della proteina bersaglio e del ligando il virtual screening viene eseguito seguendo le metodiche del docking molecolare. Sebbene diversi lavori ne documentino il successo, tale applicazione è caratterizzata da diverse limitazioni dovute all’intrinseca tendenza ad approssimare i fattori reali dell’ambiente biologico di una proteina in soluzione, quali il movimento continuo della struttura proteica nello spazio, effetto dei legami idrogeno in soluzione, entropia e forze di Van Der Waals, per citarne alcuni (28). In assenza della struttura tridimensionale della proteina bersaglio si esegue un virtual screening basato su ligando, metodica che si basa sul modellamento chimico del farmacoforo, definito come l’insieme delle sottostrutture della molecola di un farmaco necessarie all’interazione col recettore (29). Modelli farmacofori vengono generati estraendo descrittori molecolari da molecole note che legano il bersaglio di interesse, per poi confrontarli tramite il virtual screening con le librerie chimiche con lo scopo di indentificare nuovi ligandi. Recentemente l’applicazione di metodiche di DL al virtual screening ha dimostrato evidenze di superiorità rispetto ai metodi tradizionali grazie alle capacità di classificazione ed estrazione di descrittori molecolari (30).
Hit-to-lead Optimization
Lo scopo di questa fase è selezionare ulteriormente gli Hit Compounds che posseggono caratteristiche farmacodinamiche di potenza ed affinità richieste (Hit-to-Lead phase) ed ottimizzare le strutture molecolari dei composti Lead ottenuti (Lead optimization phase) affinché acquisiscano le caratteristiche farmacocinetiche e di tollerabilità indispensabili per la sperimentazione in vivo successiva (27).
Tipicamente questo processo si avvale di analisi basale sulla relazione quantitativa struttura-attività (Quantitative structure-activity relationship – QSAR), termine che si riferisce all’uso di metodi matematici per lo studio delle relazioni tra le proprietà fisico-chimiche, l’attività biologica e strutture dei composti la cui attività sperimentale è ignota. Questi modelli si basano sul principio fondamentale che molecole con proprietà chimico-fisiche simili fra loro avranno anche attività simile. I descrittori molecolari (es. peso molecolare, area, volume, momento dipolare, flessibilità, capacità di formare legami d’idrogeno, etc.) vengono estratti dai composti in studio ed analizzati sulla base del calcolo dei descrittori appartenenti ad un set di molecole di cui è nota sperimentalmente l’attività biologica (31). I primi modelli di analisi QSAR risalgono agli anni 60 dello scorso secolo (32), da allora diversi metodi di rappresentazione delle strutture chimiche e modelli matematici potenziati con tecniche computazionali di machine learning so- no stati sperimentati con successo, al punto da rendere l’analisi QSAR uno degli strumenti computazionali più utilizzati nel processo di lead optimization, soprattutto nel caso di bersagli farmacologici la cui struttura tridimensionale non è nota (33).
Ad ogni modo i modelli di QSAR basati tu modelli di ML si sono dimostrati ancora perfezionabili, manifestando problematiche tipiche come l’over fitting, particolarmente evidenti nel caso di analisi di strutture chimiche con differenze sostanziali rispetto al set di molecole note utilizzate per il training dei modelli di QSAR. I modelli tradizionali di QSAR non sono funzionali per l’analisi dei big data a ragione del volume, velocità ed eterogeneità che li caratterizza. Negli ultimi anni le tecniche di QSAR basate su DL, grazie proprio alla capacità di analisi di grandi quantità di dati non strutturati, hanno dimostrato di migliorare le performance di predizione dell’attività biologica e rappresentano un futuro promettente nelle fasi di ottimizzazione hit-to-lead (34).
Design de novo
In alternativa allo screening di librerie di composti, l’individuazione della specifica struttura chimica capace di esplicare le proprietà biologiche desiderate sui bersagli desiderati rappresenta uno dei quesiti principali nel processo di drug discovery. Il disegno di molecole de novo ottimizza questo processo utilizzando gli algoritmi predittivi di attività nell’analisi di librerie chimiche e simulando i cicli secondo la sequenza design-make-test. Questi cicli eseguiti in silico possono fornire, su base predittiva, una lista di composti candidati con le caratteristiche ricercate ab initio. Si tratta di un processo caratterizzato da notevole complessità in considerazione della stima di 1060 composti farmaceutici teoricamente possibili, un ordine di grandezza superiore al numero stimato di atomi presenti nel sistema sola- re (35). Le metodiche di DL hanno il potenziale di affrontare questa sfida attraverso la capacità di generazione automatica e ragionata di composti tramite algoritmi che cataloghino, caratterizzino e confrontino le proprietà di milioni di composti in silico, per aiutare i ricercatori a trovare, in modo rapido e conveniente, i migliori farmaci candidati per un target. L’analisi automatizzata potrebbe anche aiutare ad aprire aree di spazio chimico lasciate inesplorate o ritenute sterili. Diversi studi recenti hanno proposto modelli applicativi e mostrato prove di concetto ottimistiche (36,37). In uno di questi studi, attraverso l’apprendimento rinforzato, è stato possibile generare antagonisti del recettore dopaminergico di tipo II con un’accuratezza, in termini di bioattività delle molecole proposte, superiore al 95% (38).
Processo di sintesi
Anche se teoricamente le tecniche di chimica consentono di sintetizzare quasi qualsiasi composto desiderato, alcuni composti presentano delle difficoltà tecniche intrinseche non trascurabili. I processi di design de novo possono facilmente suggerire milioni di strutture chimiche potenzialmente funzionali, senza però fornire indicazioni sul metodo di sintesi e/o sulla prioritizzazione dei composti in base ad un criterio di fattibilità. Il piano di sintesi ideale soddisfa i requisiti di accessibilità dei costi, alta resa, assenza di reagenti pericolosi. Per soddisfare entrambe queste esigenze sin dagli anni ’60 è stato utilizzato il sistema CASP (computer-aided synthesis planning) i cui algoritmi utilizzano tipicamente due tipologie di database: il primo contenente le reazioni chimiche conosciute e il secondo i composti di partenza, quali le molecole normalmente disponibili in commercio.
Attualmente i sistemi di machine learning consentono di istruire i circuiti neuronali artificiali con regole di chimica organica, affinché essi possano proporre percorsi di sintesi appropriati e suggerire una prioritizzazione dei composti in base al criterio di fattibilità di sintesi. I sistemi di DL consentono di effettuare sia predizioni di sintesi prospettica – nelle quali i prodotti sono predetti a partire dai singoli reagenti – sia predizioni di retrosintesi, nelle quali vengono predetti sia i prodotti finali che le reazioni necessarie alla loro sintesi (39).
Un recente lavoro ha dimostrato come i sistemi artificiali possano apprendere i meccanismi di chimica organica autonomamente a partire dai database di reazioni chimiche note senza necessità di training da parte di esperti umani. In questo studio le reti neuronali di DL sono state istruite sulla base delle reazioni biochimiche pubblicate in letteratura e il sistema artificiale ha risolto il doppio delle reazioni biochimiche, circa 30 volte più velocemente rispetto ai metodi computerizzati tradizionali. Inoltre, da un’analisi in doppio cieco eseguita da chimici esperti, la cascata di reazioni proposta dai sistemi di DL è risultata in media sovrapponibile a quella riportata in letteratura. Questo dato non sorprende, alla luce del fatto che il sistema artificiale è stato istruito difatti proprio sulla base della letteratura accademica riconosciuta (40).
Valutazione in silico delle proprietà ADME/T
La safety di un composto in fase di sviluppo rappresenta certamente una delle principali cause di fallimento, con il 30% di attrition rate globale attribuibile proprio a ragioni di tossicità (41). La probabilità che un nuovo composto possa generare eventi avversi non è praticamente mai considerabile nulla, difatti anche una volta in commercio il farmaco può essere ritirato per ragioni di safety. Per tali motivi la valutazione della sicurezza di un composto sin dalle prime fasi è un aspetto della massima importanza. Generalmente la drug safety nelle fasi precliniche viene valutata attraverso processi sperimentali basati su test in vitro e in vivo. Recentemente sono stati sviluppati anche modelli di cultura cellulare 3D che permettono l’analisi su strutture tissutali organizzate e differenti popolazioni di cellule differenziate (42). Queste culture chiamate ‘organs-on-chips’ permettono di studiare nuovi composti in condizioni molto vicine al contesto biologico, ma richiedono, come per le tradizionali tecniche in vivo e in vitro, notevoli risorse in termini di costi e tempi. Al contrario le metodiche computazionali presentano numerosi vantaggi, tra i quali l’assenza delle criticità anche etiche dei modelli animali, velocità, costi contenuti e soprattutto la possibilità di analisi prima ancora della sintesi del composto. La capacità del DL di estrazione automatica delle caratteristiche molecolari ha permesso negli ultimi anni di ottenere affidabili predizioni di parametri di tossicità come la dose letale mediana, LD50, dose di una sostanza, somministrata in una volta sola, in grado di uccidere il 50% di una popolazione campione. Questo parametro, tipico indicatore di tossicità orale, è predicibile con una accuratezza >95% tramite architetture di deep learning liberamente disponibili nel web (43).
Tramite tecniche di machine learning è possibile predire i parametri di analisi farmacocinetica di un composto, come la solubilità direttamente dall’analisi computerizzata dei descrittori molecolari capaci di elaborare caratteristiche dei legami chimici e gruppi funzionali (44).
L’assorbimento di un farmaco è definito dal passaggio al circolo ematico a partire dal sito di somministrazione, fenomeno da cui ne deriva la biodisponibilità in termini farmacocinetici. I metodi computazionali hanno dimostrato di predire accuratamente la capacità di passaggio nel comparto attraverso il monostrato cellulare in vitro, espresso come coefficiente di permeabilità apparente (Papp); dimostrando una valida alternativa al modello di assorbimento intestinale basato sulle cellule Caco-2, isolate da un adenocarcinoma colon-rettale umano (45). Tale modello è un sistema in vitro ben caratterizzato che rende possibile studiare la tossicità orale di tutte quelle sostanze che vengono ingerite intenzionalmente o accidentalmente, definire il loro meccanismo di trasporto attraverso la barriera intestinale, e quindi determinare la biodisponibilità delle stesse nel sangue e nei tessuti.
La distribuzione di un farmaco invece definisce il processo di passaggio del composto dalla componente plasmatica verso l’interstizio e lo spazio intracellulare. La distribuzione allo stato stazionario (steady state) è un importante parametro farmacocinetico che esprime la distribuzione del farmaco verso i bersagli. Predire prima della sintesi questo parametro consente quindi di apportare modificazioni chimiche al composto al fine di ottimizzarne le proprietà farmacocinetiche. Attualmente le tecniche computazionali di predizione hanno dimostrato risultati incoraggianti ma soggetti a miglioramento, questo in ragione della presenza di numerose variabili proprie dell’organismo che influenzano la distribuzione del farmaco, ovviamente non prevedibili a partire dalla struttura molecolare del composto (46). A seguito della distribuzione all’interno dell’organismo il farmaco è soggetto a metabolismo, a cui può conseguire una perdita di funzione della molecola o produzione di metaboliti tossici. La predizione di questi meccanismi può guidare l’ottimizzazione ragionata della struttura molecolare al fine di ottenere l’effetto desiderato. Nella maggior parte dei casi è desiderabile una stabilità del composto che mantenga l’efficacia dopo il passaggio metabolico, mentre in altri casi, come per i corticosteroidi inalatori, risulta vantaggioso che il farmaco venga degradato a livello epatico per evitarne la presenza sistemica. Oggi sono disponibili numerose piattaforme che permettono questo tipo di analisi con risultati performanti, ad esempio è possibile, utilizzando metodiche di machine learning, stabilire la possibilità che un sito di una pic- cola molecola venga metabolizzato dalla famiglia dei citocromi CYP450 con una accuratezza globale dell’87% (47).
A seguito della distribuzione, il composto farmaceutico può essere eliminato dall’organismo direttamente o come prodotto del metabolismo; la capacità di predire questo processo è dunque influenzata dalla conoscenza del metabolismo e dei parametri di solubilità dei metaboliti. Modelli in silico hanno dimostrato di valutare e predire i meccanismi di clearance con un livello di accuratezza dell’84% (48).
Inoltre l’analisi multitasking di assorbimento, distribuzione, metabolismo, escrezione e tossicità tramite reti neuronali artificiali ha dimostrato di migliorare ulteriormente la performance dell’analisi parametri presi singolarmente (49).
Drug repurposing
Il riposizionamento dei composti definisce un processo finalizzato all’individuazione di nuove indicazioni di farmaci già approvati per la fase di sviluppo clinico utile al ridurre il tempo necessario e il rischio di fallimento nel corso della ricerca e sviluppo del farmaco (50).
Il principio alla base del riposizionamento risiede nell’eterogeneità di bersagli e dei pathways fisiopatologici a valle, in parte parzialmente noti, che uno stesso composto può perturbare ed ai quali corrispondono eventi in diversi sistemi biologici potenzialmente da esplorare.
Sebbene in linea teorica composti simili esercitino perturbazioni simili nei sistemi biologici, una gestione razionale del riposizionamento di composti già noti richiede una conoscenza approfondita dei network intra ed extra cellulari, quali i network di regolazione genica, metabolici, cascate del segnale, interazioni proteina-proteina, interazioni farmaco-bersaglio, farmaco-farmaco, farmaco-malattia, farmaco-bersagli non desiderati e network biochimici malattia-malattia (51).
Le informazioni di una singola rete di segnali biomolecolari sono spesso limitate e parziali, rendendo quindi necessario integrare più reti precedentemente individuate separatamente. Tramite questo approccio è stato possibile ipotizzare che molecole già note per altre indicazioni come l’alendronato, il telmisartan e la clorpropamide, rispettivamente un bisfosfonato, un sartano e un ipoglicemizzante orale, avessero la capacità di inibire le ciclossigenasi. Tale ipotesi è stata successivamente confermata sperimentalmente confermando l’effetto antinfiammatorio delle molecole (52).
Conferme sperimentali di questo tipo consentono a molecole su cui sono state investite risorse ingenti e di cui si conoscono i dati preliminari di safety ed ef cacy di essere studiate e modificate per verificarne l’efficacia su altri target, accelerando ed ottimizzando quindi il processo di selezione di nuovi composti.
Sistemi di ML hanno dimostrato di poter rivalutare e suggerire una nuova sperimentazione in altre indicazioni in composti la cui sperimentazione clinica è stata interrotta. Uno di questi composti è il bavisant, inibitore dei recettori istaminergici H3 inizialmente in sviluppo in fase II per lo studio del disturbo da deficit di attenzione e iperattività, la cui sperimentazione è stata ampliata per il trattamento dei pazienti con malattia di Parkinson (53).
Esperienza nell’industria del farmaco
Per superare i limiti di risorse, tempi ed inefficienza tipici dello sviluppo di nuovi composti molte compagnie farmaceutiche stanno esplorando e, in alcuni casi, applicando l’utilizzo di intelligenza artificiale nelle diverse fasi della R&S del farmaco. In anni recenti l’applicazione di queste tecnologie al settore farmaceutico nasce dal trasferimento tecnologico da start-up specializzate che collaborano a vario titolo con realtà consolidate nel settore. Una recente stima, in continuo aggiornamento, di questo fenomeno documenta ben 129 startups (54) impegnate nell’utilizzo di Artificial Intelligence nel Drug Discovery le quali forniscono alle compagnie farmaceutiche una serie di servizi classificabili nelle diverse finalità quali:
1) aggregazione e sintesi di informazioni;
2) comprensione dei meccanismi di patologia;
3) generazione di dati e modelli;
4) repurposing di farmaci esistenti;
5) generazione di nuovi composti candidati;
6) validazione e ottimizzazione dei composti candidati;
7) drug design;
8) disegno sperimentazioni precliniche;
9) esecuzione di sperimentazioni precliniche;
10) disegno di trials clinici;
11) reclutamento di pazienti per sperimentazioni cliniche;
12) ottimizzazione dei trials clinici;
13) pubblicazione di dati.
Sono attualmente almeno 30 le compagnie farmaceutiche che hanno avviato programmi di intelligenza artificiale per la R&S del farmaco, sia all’interno della propria organizzazione sia attraverso collaborazioni con startup(55). La grande opportunità offerta da questi nuovi approcci in associazione alla necessità di condivisione di dati ha portato anche a collaborazioni tra privati e collaborazioni miste pubblico-privato come nel caso della piattaforma Open Targets Validation Platform liberamente accessibile (https://www.targetvalidation.org). Quest’ultima è una piattaforma di integrazione e visualizzazione dei dati che fornisce evidenze sull’associazione di bersagli farmacologici noti e potenziali con malattie.
Ogni target farmacologico è collegato a una malattia utilizzando dati a livello genomico provenienti da un’ampia gamma di fonti di dati, permettendo ai ricercatori di interrogare la piattaforma ed ottenere immediatamente informazioni su oltre 20 mila target che, sulla base di oltre 3 milioni di associazioni, sono correlati a oltre 10 mila patologie.
Conclusioni
In conclusione, l’intelligenza artificiale ha la capacità di intervenire potenziando la capacità di identificare nuovi target potenziali, scoprire nuove molecole, predire il funzionamento dei composti e la tossicità, creare un utilizzo personalizzato dei composti sulla base di marcatori genici. La sua maggiore aspettativa interessa la riduzione della principale criticità della R&S del farmaco, ovvero il tasso di fallimento nello sviluppo clinico. Difatti anche recenti valutazioni retrospettive trasversali della struttura dei processi industriali tradizionali e la successiva applicazione delle “lessons learned” hanno prodotto un tasso di successo dei composti candidati non superiore al 19% (56).
Ma prima che qualsiasi tecnologia di intelligenza artificiale possa dominare i processi di scoperta e sviluppo del farmaco è necessario che uno o più progetti mantengano le proprie promesse. Esistono difatti, ad oggi, diverse criticità riguardo l’applicazione di questa tecnologia in questo settore, come la disponibilità di grandi quantità di dati affidabili utili al training della macchina sul quale si basa la performance della mansione che viene poi richiesta alla stessa; inoltre i meccanismi di deep learning sono in parte ancora sconosciuti. Così come i meccanismi neurofisiologici dell’encefalo umano sono difficili da comprendere dall’esterno, allo stesso modo i network computazionali sono troppo complessi affinché il ricercatore comprenda la profonda natura di quanto suggerito dalla macchina. A questo si aggiunge la mancanza di linea guida e protocolli condivisi sulle modalità di training e correzioni della macchina necessari a questo scopo durante il processo stesso.
Non è la prima volta infatti che le compagnie farmaceutiche si affidano a soluzioni high tech per aumentare la produttività di R&S. L’introduzione dello high throughput screening, con l’uso di robot per testare rapidamente milioni di molecole, ha generato enormi quantità di composti nei primi anni del 2000, ma non è ancora riuscita a risolvere le inefficienze nel processo di ricerca. Se nel prossimo futuro, come atteso, la tecnologia dell’intelligenza artificiale saprà convogliare l’analisi automatizzata ed integrata di tutti i dati necessari allo sviluppo di un farmaco (docking molecolare, simulazioni di chemioformatica, bioinformatica, dati di -omica) è plausibile che si potrà assistere ad una vera rivoluzione di come la ricerca mette a disposizione nuovi composti per affrontare i bisogni di salute della popolazione mondiale.
Bibliografia
- DiMasi, JA, et al. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ 2016; 47: 20-33.
- Wong CH, et al. Estimation of clinical trial success rates and related parameters. Biostatistics 2019; 20: 273-86.
- United Nations, Department of Economic and Social Affairs, Population Division (2017). World Population Prospects: The 2017 Revision,
Key Findings and Advance Tables. ESA/P/WP/248. - Griggs RC, et al. Clinical research for rare disease: opportunities, challenges, and solutions. Mol Genet Metab 2009; 96: 20-6.
- Harrison RK. Phase II and phase III failures: 2013-2015.
Nat Rev Drug Discov 2016; 15: 817-8. - Duch W, et al. Artificial intelligence approaches for rational drug design and discovery. Curr Pharm Des 2007; 13: 1497-1508.
- Sliwoski G, et al. Computational methods in drug discovery.
Pharmacol Rev 2014; 66: 334-95. - Gantz J, Reinsel D. The digital universe decade–are you ready (2010). http://www. emc. com/collateral/analyst-reports/idcdigital-universe-are-you-ready. pdf (2012).
- Ekins S. The next era: Deep learning in pharmaceutical research. Pharm Res 2016; 33: 2594-603.
- Chen H, et al. The rise of deep learning in drug discovery. Drug Discov Today 2018; 23: 1241-50.
- Chen Y, et al. IBM Watson: how cognitive computing can be applied to big data challenges in life sciences research. Clin Ther 2016; 38: 688-701.
- Topol EJ. High-performance medicine: the convergence of human and artificial intelligence. Nature Med 2019; 25: 44.
- Korotcov A, et al. Comparison of deep learning with multiple machine learning methods and metrics using diverse drug discovery data sets. Mol Pharm 2017; 14: 4462-75.
- Hunter P. The in ammation theory of disease: The growing realization that chronic in ammation is crucial in many diseases opens new avenues for treatment. EMBO Rep 2012; 13: 968-70.
- Gerstein HC, et al; Action to Control Cardiovascular Risk in Diabetes Study, Group. Effects of intensive glucose lowering in type 2. N Eng J Med 2008; 358: 2545-59.
- Nelson MR, et al. The support of human genetic evidence for approved drug indications. Nature Genet 2015; 47: 856.
- King EA, et al. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. BioRxiv 2019: 513945. doi: https://doi.org/10.1101/513945.
- Lombardo R, Priami C. Graphical modeling meets systems pharmacology. Gene Regul Syst Biol 2017; 11: 1177625017691937. doi: 10.1177/1177625017691937.
- Lauria M, et al. SCUDO: a tool for signature-based clustering of expression pro les. Nucleic Acids Res 2015; 43: W188-W92.
- Nassiri I, et al. Systems view of adipogenesis via novel omics-driven and tissue-speci c activity scoring of network functional modules. Sci Rep 2016; 6: 28851.
- Spencer M, et al. A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Trans Comput Biol Bioinf2015; 12: 103-12.
- Higueruelo AP, et al. Protein-protein interactions as druggable targets: recent technological advances. Curr Opin Pharmacol 2013; 13: 791- 6.
- Shin W-H, et al. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods 2017; 131: 22-2.
- Valkov E, et al. Targeting protein–protein interactions and fragment-based drug discovery. In: Fragment-Based Drug Discovery and X-Ray Crystallography. Springer, Berlin, Heidelberg, 2011; 145-79.
- Du T, et al. Prediction of residue-residue contact matrix for protein-protein interaction with Fisher score features and deep learning. Methods 2016; 110: 97-105.
- Torng W, Russ BA. 3D deep convolutional neural networks for amino acid environment similarity analysis. BMC Bioinformatics 2017; 18: 302.
- Hughes JP, et al. Principles of early drug discovery. Br J Pharmacol 2011; 162: 1239-49.
- Chen Y-C. Beware of docking! Trends Pharmacol Sci 2015; 36: 78-95.
- Wermuth CG, et al. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl Chem 1998; 70: 1129-43.
- Unterthiner T, et al. Deep learning as an opportunity in virtual screening. Proceedings of the deep learning workshop at NIPS. 2014; 27.
- Esposito EX, et al. Methods for applying the quantitative structure-activity relationship paradigm. Methods Mol Biol 2004; 275: 131-214.
- Corwin H, Fujita T. ρ-σ-π Analysis. A method for the correlation of biological activity and chemical structure. J Am Chem Soc 1964; 86: 1616-26.
- Wang T, et al. Quantitative structure–activity relationship: promising advances in drug discovery platforms. Expert Opin Drug Discov 2015; 10: 1283-300.
- Ma J, et al. Deep neural nets as a method for quantitative structure-activityrelationships. J Chem Inf Model 2015; 55: 263-74.
- Mullard A. The drug-maker’s guide to the galaxy. Nature 2017; 549: 445-7.
- Gómez-Bombarelli R, et al. Automatic chemical design using a data-drivencontinuous representation of molecules. ACS Cent Sci 2018; 4: 268-76.
- Popova M, et al. Deep reinforcement learning for de novo drug design. Sci Adv 2018; 4: eaap7885.
- Olivecrona M, et al. Molecular de-novo design through deep reinforcement learning. J Cheminform 2017; 9: 48.
- Coley CW, et al. Machine learning in computer-aided synthesis planning. Acc Chem Res 2018; 51: 1281-9.
- Segler MHS, et al. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018; 555: 604-10.
- Giri S, Bader A. A low-cost, high-quality new drug discovery process using patient-derived induced pluripotent stem cells. Drug Discov Today 2015; 20: 37-49.
- Huang R, et al. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat Commun 2016; 7: 10425.
- Xu Y, et al. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chemical Inf Model 2017; 57: 2672-85.
- Coley CW, et al. Convolutional embedding of attributed molecular graphs for physical property prediction. J Chemical Information Model 2017; 57: 1757-72.
- Wang S, et al. ADMET evaluation in drug discovery. 16. Predicting hERG blockers by combining multiple pharmacophores and machine learning approaches. Mol Pharm 2016; 13: 2855-66.
- Lombardo F, Yankang J. In silico prediction of volume of distribution in humans. Extensive data set and the exploration of linear and nonlinear methods coupled with molecular interaction elds descriptors. J Chemical Inf Model 2016; 56: 2042-52.
- Zaretzki J, et al. XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J Chemical Inf Model 2013; 53: 3373-83.
- Lombardo F, et al. Clearance mechanism assignment and total clearance prediction in human based upon in silico models. J Med Chem 2014; 57: 4397-405.
- Kearnes S, et al. Modeling industrial ADMET data with multitask networks.arXiv preprint 2016; arXiv:1606.08793.
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Reviews Drug Discov 2004; 3: 673 – 83.
- Lot Shahreza M, et al. A review of network-based approaches to drug repositioning. Brief Bioinform 2017; 19: 878-92.
- Luo Y, et al. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun 2017; 8: 573.
- https://clinicaltrials.gov/ct2/show/NCT03194217?term=bavisant&rank=2. April 2019.
- Smith S. 129 startups using arti cial intelligence in drug discovery. BenchSci (April 2019). https://blog.benchsci.com/startups-using-artificial-intelligence-in-drug-discovery.
- Smith S. 30 pharma companies using arti cial intelligence in drug discovery. The BechSci blog (April 2019). https://blog.benchsci.com/pharma-companies-using-artificial-intelligence-in-drug-discovery.
- Morgan P, et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov 2018; 17: 167-81.



