Alice Ravizza1, Enrico Caiani2, Eugenio Santoro3, Silvia Stefanelli4, Federico Sternini1
1Use-Me-D,Torino
2Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano
3Laboratorio di Informatica Medica, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano
4Studio Legale Stefanelli & Stefanelli, Bologna
Tendenze nuove, Numero Speciale n.01 2021; 15-36: DOI: 10.32032/TENDENZENS20210103.PDF
 Scarica Pdf
Scarica Pdf
1. Inquadramento di tipo regolatorio
1.1. La nozione di dispositivo medico nella Direttiva 93/42/CEE
Il dispositivo medico viene definito tecnicamente nell’ordinamento europeo con la Direttiva 93/42/CEE attuata in Italia dal Decreto Legislativo 46/97.
L’articolo 1 lettera a) della Direttiva 93/42/CEE nella versione originaria riportava la seguente definizione di dispositivo medico:
“qualsiasi strumento, apparecchio, impianto, sostanza o altro prodotto, utilizzato da solo o in combinazione, compreso il software informatico impiegato per il corretto funzionamento e destinato dal fabbricante ad essere impiegato nell’uomo per:
• diagnosi, prevenzione, controllo, terapia o attenuazione di una malattia;
• diagnosi, controllo, terapia, attenuazione o compensazione di una ferita o di un handicap;
• studio, sostituzione o modifica dell’anatomia o di un processo fisiologico;
• intervento sul concepimento;
la cui azione principale voluta nel o sul corpo umano non sia conseguita con mezzi farmacologici né immunologici né mediante metabolismo, ma la cui funzione possa essere assistita da questi mezzi”.
Alla luce di questa definizione il software rientrava pertanto nella nozione di dispositivo medico non in via autonoma, ma solo ove impiegato per il corretto funzionamento del dispositivo stesso.
La versione originaria del 1993, dunque, intendeva regolare il software cosiddetto “embedded” destinato a permettere il funzionamento e a fornire una interfaccia ai dispositivi elettromedicali. Il software cosiddetto “stand-alone” non è descritto.
1.2. L’allargamento al software della nozione di dispositivo medico nella Direttiva 2007/47/CEE
Nel 2007 il legislatore comunitario interviene con una nuova Direttiva (2007/47/CEE – MDD) ad allargare la nozione di dispositivo medico, stabilendo che il software può essere di per sé un dispositivo medico.
La portata di tale modifica emerge con chiarezza nel Considerando 6 della Direttiva stessa ove si stabilisce che:
“Occorre chiarire che un software è di per sé un dispositivo medico quando è specificamente destinato dal fabbricante ad essere impiegato per una o più delle finalità mediche stabilite nella definizione di dispositivo medico. Anche se utilizzato in un contesto sanitario, il software generico non è un dispositivo medico”.
Il successivo Considerando 20 sancisce poi che:
“Considerata l’importanza crescente del software nel settore dei dispositivi medici, come software indipendente (stand-alone) oppure come software incorporato in un dispositivo, un requisito essenziale dovrebbe essere la validazione del software secondo lo stato dell’arte”.
La nozione di dispositivo medico viene quindi riformulata nel seguente modo:
“qualunque strumento, apparecchio, impianto, software, sostanza o altro prodotto, utilizzato da solo o in combinazione, compresi gli accessori tra cui il software destinato dal fabbricante ad essere impiegato specificamente con finalità diagnostiche e/o terapeutiche e necessario al corretto funzionamento del dispositivo stesso, destinato dal fabbricante ad essere impiegato sull’uomo a fini di:
• diagnosi, prevenzione, controllo, trattamento o attenuazione di malattie;
• diagnosi, controllo, trattamento, attenuazione o compensazione di una ferita o di un handicap;
• studio, sostituzione o modifica dell’anatomia oppure di un processo fisiologico;
• controllo del concepimento;
che non eserciti nel o sul corpo umano l’azione principale cui è destinato con mezzi farmacologici, immunologici o mediante processi metabolici, ma la cui funzione possa essere coadiuvata da tali mezzi”.
Dal 2007 in poi il legislatore ha dunque sancito che il software può essere considerato un dispositivo medico sia quando è incorporato nel dispositivo (embedded) sia quando opera a sé stante (stand-alone).
Successivamente, il software come dispositivo medico stand-alone (anche detto Software as a Medical Device – SaMD) è descritto nella linea guida internazionale IMDRF/SaMD WG/N10FINAL:2013 con i seguenti termini:
“The term “Software as a Medical Device” (SaMD) is defined as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device”.
1.3. Il Regolamento 2017/745/CE sui dispositivi medici
Nel 2017 veniva poi approvato il nuovo Regolamento UE 2017/745/CE sui dispositivi medici (cosiddetto MDR – Medical Device Regulation, nel seguito anche “il Regolamento”). Tale Regolamento, pubblicato sulla Gazzetta Ufficiale dell’Unione Europea in data 5 maggio 2017, avrebbe dovuto abrogare l’attuale Direttiva e sarebbe dovuto entrare in vigore in data 26 maggio 2020. Tuttavia, nel mese di aprile 2020, a causa della pandemia COVID-19, il Parlamento Europeo ha derogato l’entrata in vigore del nuovo Regolamento ed esteso la copertura dell’attuale Direttiva di un anno, rendendo la data di piena applicazione il 26 maggio 2021. Occorre notare che il Regolamento, al contrario della Direttiva, è un vero e proprio atto legislativo vincolante in tutte le sue parti, che non ha bisogno di essere recepito, e che obbliga gli Stati membri a rispettarlo per intero.
Il nuovo MDR ha esteso la definizione di dispositivo medico, includendo predizione e prognosi tra le finalità considerate, andando così potenzialmente a includere software di calcolo di indici di rischio.
Per quanto concerne l’applicazione della nuova disciplina ai software, in particolare, è importante evidenziare che l’articolo 2 punto 28 del MDR definisce che l’immissione sul mercato è da intendersi come “la prima messa a disposizione di un dispositivo sul mercato dell’Unione, escludendo dispositivi usati in studi clinici”.
Il successivo articolo 5 sancisce che “Un dispositivo può essere immesso sul mercato o messo in servizio solo se è conforme al presente Regolamento qualora sia debitamente fornito e correttamente installato, oggetto di un’adeguata manutenzione e utilizzato conformemente alla sua destinazione d’uso”.
Il Considerando 19, cercando di meglio definire quando il software sia un dispositivo medico, così stabilisce: “è necessario precisare che il software destinato dal fabbricante a essere impiegato per una o più delle destinazioni d’uso mediche indicate nella definizione di dispositivo medico si considera un dispositivo medico, mentre il software destinato a finalità generali, anche se utilizzato in un contesto sanitario, o il software per fini associati allo stile di vita e al benessere non è un dispositivo medico. La qualifica di software, sia come dispositivo sia come accessorio, è indipendente dall’ubicazione del software o dal tipo di interconnessione tra il software e un dispositivo”. Quindi i software cosiddetti di wellness, cioè di semplice tracciatura dello stile di vita, non corrispondono alla definizione di dispositivo medico perché non destinati alla “diagnosi, prevenzione, monitoraggio, previsione, prognosi, trattamento o attenuazione di malattie”, mentre software che consentono di tracciare e raccogliere dati relativi alla vita quotidiana con finalità di prevenzione primaria o secondaria possono rientrare nella definizione di dispositivo medico.
Le responsabilità del fabbricante vengono definite nell’Articolo 10 del Regolamento e comprendono tra i principali:
• la necessità di dotarsi di un sistema per la gestione del rischio, che comprenda tutte le fasi del ciclo di vita del prodotto medicale, dalla progettazione alla dismissione, con nuova e più approfondita necessità di valutazione dei dati post-certificazione;
• l’obbligo di effettuare una valutazione clinica per la determinazione del beneficio del prodotto.
L’Allegato I del Regolamento comprende una lista di Requisiti Essenziali, che devono essere presi in considerazione dal fabbricante per tutti i dispositivi, qualsiasi sia la classe di rischio. Per la dimostrazione di conformità ai Requisiti applicabili, il fabbricante può adottare diverse strategie (analisi, test o procedure): tipicamente, la conformità a norme internazionali e a norme armonizzate, quando disponibili, viene considerata la strategia più adeguata.
Nel caso delle Terapie Digitali (Digital Therapeutics – DTx), le norme internazionali più adeguate sono rispettivamente la ISO 13485 e la EN ISO 14971 per gli approcci generali, seguite dalla IEC 62304 e IEC 62366 per gli aspetti specificatamente legati al ciclo di vita e alla usabilità.
1.4. Le classi di rischio e ilsoftware dispositivo medico
Per poter identificare la classe del dispositivo medico, è necessario analizzarne l’uso previsto ed il relativo rischio intrinseco. L’identificazione della classe di rischio del dispositivo consente di individuare la strategia e la procedura necessarie per dimostrare la conformità del dispositivo alla Direttiva o al Regolamento. Queste procedure sono disegnate in maniera tale da allocare le risorse degli enti notificati e dei fabbricanti così che dispositivi ad alto rischio richiedano verifiche più rigorose, mentre dispositivi a rischio inferiore possano richiedere verifiche meno dispendiose, senza compromettere la sicurezza del paziente.
I dispositivi medici sono classificati in quattro classi di rischio crescente:
Classe I: dispositivi meno critici, quali la gran parte di quelli non attivi e non invasivi.
All’interno di detta classe sono individuabili tre sottoclassi:
Classe Is: dispositivi di classe I forniti allo stato sterile
Classe Im: dispositivi di classe I che svolgono una funzione di misura
Classe Ir: dispositivi di classe I che possono essere riprocessati (nuova classe introdotta con il MDR).
Classe IIa: dispositivi a rischio medio, quali alcuni dispositivi non attivi (invasivi e non) e dispositivi attivi che interagiscono con il corpo in maniera non pericolosa
Classe IIb: dispositivi a rischio medio/alto, quali alcuni dispositivi non attivi (specie invasivi) e i dispositivi attivi che interagiscono con il corpo in maniera pericolosa
Classe III: dispositivi ad alto rischio, quali gran parte dei dispositivi impiantabili, quelli contenenti farmaci o derivati animali, ed alcuni dispositivi che interagiscono con le funzioni di organi vitali.
Per il software, sia nella Direttiva che nel Regolamento, devono essere applicate le regole di classificazione relative ai dispositivi attivi. Inoltre, nel Regolamento sono state definite delle regole specifiche proprio per i software, colmando una lacuna normativa presente nella Direttiva.
Vengono qui richiamate sia le regole di classificazione presenti nella Direttiva che quelle presenti nel Regolamento, tenendo presente che le regole nella Direttiva verranno sostituite da quelle nel Regolamento, con un importante impatto: nella ri-classificazione secondo il Regolamento, infatti, molti prodotti software attualmente in classe I secondo Direttiva vedranno innalzata la propria classe di rischio.
Nella Direttiva, le regole di classificazione sono elencate in Allegato IX e quelle rilevanti per i software sono le numero 9, 10, 11 e 12. Tali regole sono state ampiamente descritte e commentate nella MEDDEV 2.1/6 July 2016 e nella MEDDEV 2. 4/1 Rev. 9 June 2010 di cui di seguito si riporta un estratto.
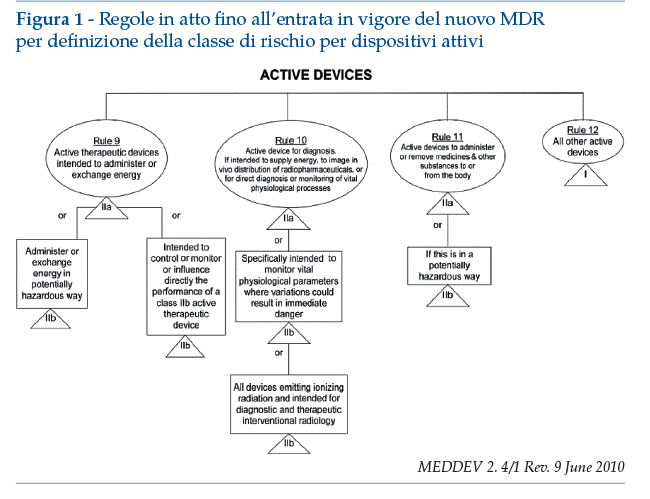
Le nuove regole di classificazione, previste nel Regolamento 2017/745/CE, sono in quest’ultimo elencate nell’Allegato VIII, dove la regola specifica sul software (regola 11) recita che:
“Il software destinato a fornire informazioni utilizzate per prendere decisioni a fini diagnostici o terapeutici rientra nella classe IIa, a meno che tali decisioni abbiano effetti tali da poter causare:
• il decesso o un deterioramento irreversibile delle condizioni di salute di una persona, nel qual caso rientra nella classe III, o
• un grave deterioramento delle condizioni di salute di una persona o un intervento chirurgico, nel qual caso rientra nella classe IIb.
Il software destinato a monitorare i processi fisiologici rientra nella classe IIa, a meno che sia destinato a monitorare i parametri fisiologici vitali, ove la natura delle variazioni di detti parametri sia tale da poter creare un pericolo immediato per il paziente, nel qual caso rientra nella classe IIb.
Tutti gli altri software rientrano nella classe I.”
In questa fase di transizione verso l’applicazione del nuovo Regolamento, vi è un evidente bisogno di approfondire le diverse classificazioni e come queste siano tra loro interconnesse, poiché tutte hanno un impatto sulle successive attività. A tale proposito la Commissione Europea ha recentemente pubblicato un documento di indirizzo, nella forma di linea guida, specificamente relativo agli aspetti regolatori del software in MDR e IVDR (MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR), così da guidare nella verifica di quando un software ricade sotto la definizione di dispositivo medico, e nella successiva determinazione della corrispondente classe di rischio.
La classe di marcatura CE, che viene definita interpretando le regole di certificazione previste nel Regolamento, dipende dall’uso previsto del software (quando esso funziona in modo coerente con quanto previsto dal fabbricante in condizioni di uso normale), ed ha un impatto sulle modalità di marcatura CE e di immissione in commercio, in particolare sul coinvolgimento di un Ente Notificato. Il coinvolgimento dell’Ente Notificato è previsto per dispositivi di classe di rischio IIa o superiore (quindi per tutti i software che sono dispositivi medici, con il nuovo Regolamento).
La classe deve essere dunque determinata analizzando l’uso previsto del software, che può essere descritto in termini di:
• popolazione di pazienti target
• utilizzatori previsti (professionali, non professionali, pazienti)
• performance clinica
• beneficio clinico atteso.
In maniera sintetica la classe può essere così descritta, per un software stand-alone dispositivo medico:

1.5. Classificazione del software dispositivo medico:
considerazioni e proposta
La Linea Guida MDCG 2019/11 precedentemente citata esplicita in modo particolare il requisito che il software eserciti la sua funzione per il beneficio di un singolo individuo, mentre esclude esplicitamente software ad uso di ricerca epidemiologica o ad uso di generica linea guida o protocollo clinico non individualizzato. In base a quanto dichiarato nella Linea Guida, si deduce facilmente che un software destinato a gestire il percorso diagnostico e terapeutico personalizzato di un singolo individuo sia da considerare un “dispositivo medico solo software” o MDSW. Il breve accenno ad un software di pianificazione terapeutica, nel Decision step 4 del paragrafo 3.3, sembra essere nella Linea Guida l’unico riferimento al fatto che un software possa svolgere un atto terapeutico. Nello specifico, la Linea Guida commenta il testo della regola 11 di MDR, già piuttosto chiaro ed esplicito, in questo modo:
“The text of Rule 11 can be divided into what are essentially three sub-rules that are applied depending on the intended use/purpose of the MDSW:
11a: (3 first paragraphs of Rule 11) intended to provide information which is used to take decisions with diagnostic or therapeutic purposes;
11b: (Paragraph 4 of Rule 11) intended to monitor physiological processes or parameters;
11c: (Paragraph 5 of Rule 11) all other uses”.
La valutazione che si può estrarre sulla base dei pochi riferimenti specifici presenti nella Linea Guida è che gli autori non abbiano approfondito la possibilità che il software eserciti un atto terapeutico, mentre si esplicitano chiaramente le possibili funzioni di
• analisi di dati allo scopo di fornire informazione, ad un essere umano o a un altro software, in modo che queste informazioni siano usate per prendere decisioni cliniche
• monitoraggio.
Addirittura nella Linea Guida si legge che (paragrafo 4.2.1)
“The wording “intended to provide information” which is used to take decisions with diagnosis or therapeutic purposes” describes, in very general terms, the “mode of action” which is characteristic of all MDSW”.
Sembra dunque che il legislatore non evidenzi in modo esplicito la possibilità che sia il software stesso ad ottenere l’effetto terapeutico, poiché tutti i software medical device hanno come destinazione di uso quella di fornire informazione (elaborata da input ad output).
Inoltre, dovrebbe essere notato che nemmeno nei molti esempi forniti nella Linea Guida, su specifici software e sulla loro classificazione, sono presenti cenni alla possibilità che un software possa essere utilizzato per terapia.
Unica eccezione, un esempio di “Cognitive therapy MDSW that includes a diagnostic function” che è classificato in classe III se la funzione diagnostica è in closed loop, mentre in classe IIa se la funzione diagnostica fornisce informazioni al medico. Nemmeno in questo caso comunque la funzione di terapia viene considerata esplicitamente come criterio di classificazione. Si cita anche l’esempio di software diagnostico “Diagnostic MDSW intended for scoring depression based on inputted data on a patient’s symptoms (e.g. mood, anxiety) should be classified as class IIb under Rule 11(a)”.
Di conseguenza, un software che abbia come uso previsto quello di fornire una terapia, ad esempio una terapia riabilitativa o cognitivo-comportamentale, potrebbe erroneamente essere fatto ricadere nel paragrafo “all other uses” ed essere classificato in classe I.
Questa interpretazione delle regole di classificazione porterebbe al rischio di classificare tutti i MDSW digital therapeutics (DTx), in classe I. Riteniamo che questa interpretazione sia una rapida ma pericolosa scorciatoia, perché porterebbe a classificare erroneamente in bassa classe di rischio prodotti software medicali molto diversi tra loro, tra cui alcuni con uso previsto di grande impatto sulla salute del paziente.
Si dovrebbe invece notare che farmaci che incorporano, in modo integrale o non integrale, un dispositivo medico (quindi SW), sono definiti come Drug-Device Combinations (DDC). Se in modo integrale, ricadono sotto il secondo sotto-paragrafo degli artt. 1(8) and 1(9) della MDR:
“1. Devices that when placed on the market or put into service incorporate, as an integral part, a substance that, if used separately, would be considered as a medicinal product, provided that the action of the substance is principal (Article 1(8) MDR). 2. Devices intended to administer a medicinal product, where they form a single integral product intended exclusively for use in the given combination and which is not reusable (Article 1(9) MDR). Typically, these devices have measuring, metering or delivery functions”.
Il documento di riferimento, relativamente alle drug-device combinations è attualmente in forma di draft2.
2 29 May 2019 EMA/CHMP/QWP/BWP/259165/2019. Committee for Medicinal Products for Human Use (CHMP). Guideline on the quality requirements for drug-device combinations
Un possibile approccio per la classificazione di DTx stand-alone, è la lettura del testo attualmente previsto, oltre ad alcuni utili riferimenti presenti nella tabella sotto riportata (tabella 2), tratta dalla Linea Guida dell’International Medical Devices Regulators Forum (IMDRF “Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations”), anche se fornita “intended for illustrative purposes only”. Questa tabella è di fatto l’unico punto nell’intero documento in cui si citi esplicitamente la parola “treat”.
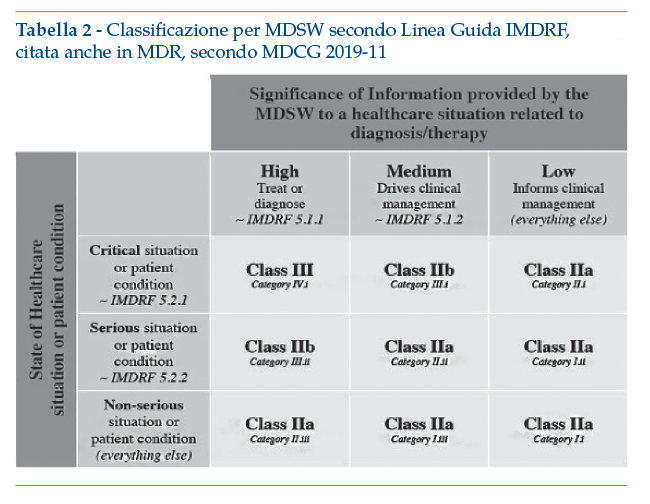
In essa, un software terapeutico si classifica in classe III, IIb oppure IIa a seconda della criticità della condizione da trattare. La classe I non è mai prevista in tabella, e anche per il livello meno critico, corrispondente alle condizioni ritenute “non serie”, si propone la classe IIa.
Di conseguenza, integrando le diverse indicazioni presenti sia nella tabella di conversione da linea guida internazionale IMDRF che nelle regole di classificazione del regolamento europeo MDR, il software terapeutico ricadrebbe sempre in una classe IIa o superiore.
Le ricadute dal punto di vista regolatorio sarebbero importantissime, anzi cruciali. Infatti, solo per la classe I è previsto che il dispositivo possa essere marcato CE e commercializzato grazie ad una “auto-dichiarazione” del fabbricante. In tutti gli altri casi, secondo Direttiva e Regolamento, per le classi IIa e superiori, è previsto un intervento di un Ente Notificato nel processo di certificazione.
Riteniamo dunque di grande interesse che gli autori delle linee guida forniscano esempi specifici per le DTx, quando applicate ai software con funzione terapeutica. Inoltre si ritiene opportuno che in tale linea guida e/o negli esempi esplicativi sia ulteriormente commentata la relazione tra la gravità della condizione da trattare con un software terapeutico e la classe di rischio, integrando gli esempi già presenti nell’Annex IV.
La classificazione delle DTx in classi IIa, IIb e III in base alla gravità della condizione da trattare ed al rischio connesso consente una buona coerenza tra i requisiti regolatori già previsti per software diagnostici e terapeutici. Infatti, la regola 11 di MDR prevede classi medio-alte di rischio per software di monitoraggio e diagnosi, innalzando anche la classe in base al livello di criticità del monitoraggio o diagnosi.
Si propone di seguito un modello per esempi di classificazione del software medical device con finalità terapeutica, che consenta di esplicitare al meglio le peculiarità delle DTx.
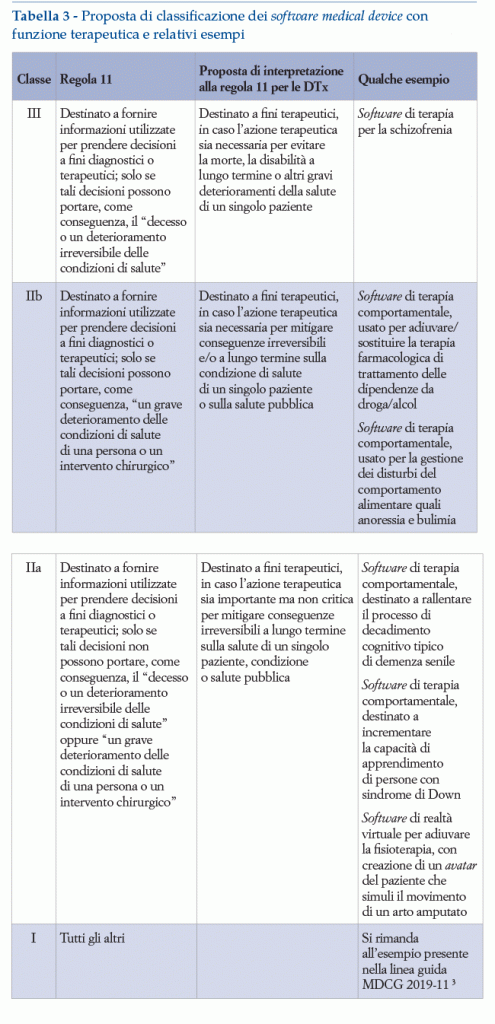
1.6. Obblighi regolatori
Come tutti i dispositivi medici, le DTx devono rispettare i requisiti essenziali previsti dalla attuale Direttiva, presenti anche nel nuovo Regolamento ma con importanti novità, soprattutto relativamente agli obblighi di dimostrazione di beneficio clinico.
Gli obblighi dei fabbricanti si possono riassumere con tre parole chiave: sicurezza, beneficio, qualità. La dimostrazione di aver adempiuto a questi obblighi avviene attraverso la applicazione di standard internazionali.
• Sicurezza
Per la dimostrazione di sicurezza, gli sviluppatori di DTx possono fare riferimento allo standard IEC 62304, la cui versione in vigore risale al 2006 ma che è attualmente in fase di ampia e significativa revisione. Questo standard definisce i metodi per progettare, sviluppare, testare il software e gestirne l’intero ciclo di vita, compresi gli aggiornamenti di versione.
L’applicazione di questo standard non può prescindere dalla contemporanea applicazione dello standard ISO 14971, relativo alla gestione del rischio, che impatta in particolare le attività di progettazione, test e aggiornamento versione. Il risultato della applicazione di questo standard consente di dimostrare che il software è privo di difetti strutturali che possano costituire un rischio per il paziente o inficiare il corretto funzionamento del software, espresso in termini di performance tecnica.
La classe di sicurezza software, definita secondo le regole descritte dalla norma tecnica IEC 62304, dipende dalla gravità del danno causato da un eventuale guasto del software stesso. Questa classe determina il rigore con cui deve essere eseguito e controllato il processo di progettazione e convalida a cura del fabbricante.
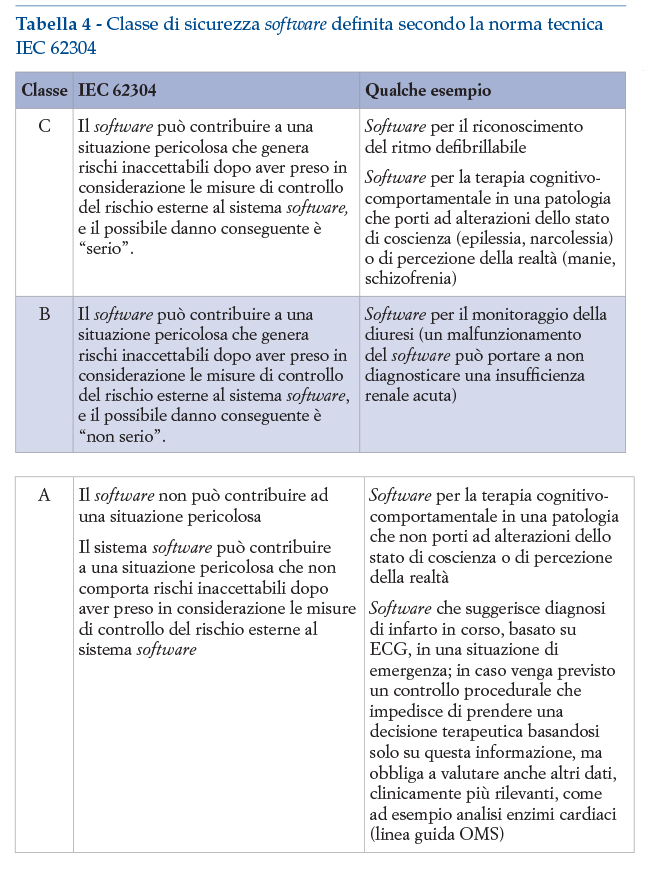
Si ritiene importante far notare come, per la conformità al requisito essenziale 17 del MDR, il fabbricante sia tenuto ad applicare principi di gestione dell’intero ciclo di vita del software: questi principi sono chiaramente elencati nella norma IEC 62304, nella sua versione EN 62304 che è una norma armonizzata. La applicazione delle norme armonizzate permette presunzione di conformità ai requisiti essenziali: questa norma dunque diventa uno strumento tecnico di grandissima rilevanza per la conformità al MDR.
È importante sottolineare che la classificazione in classi di rischio da I a III secondo MDR e la classificazione in classi di rischio da A a C secondo IEC 62304 non sono formalmente correlate. La classificazione deve essere stabilita dal fabbricante caso per caso.
• Beneficio
Per la dimostrazione di beneficio, gli sviluppatori di DTx hanno come riferimento lo standard ISO 14155, relativo alle buone pratiche di sperimentazione clinica per i dispositivi medici. L’applicazione di questo standard prevede la dimostrazione che il software sia in grado di ottenere, nella popolazione campione, un beneficio clinico significativamente superiore al rischio potenziale di utilizzo. Si passa, dunque, da indicatori di prestazione e sicurezza tecnica a indicatori di beneficio e sicurezza clinica.
Tipicamente, questa relazione viene studiata in studi di piccola numerosità, per quanto adeguata a una buona valutazione statistica. Questi studi dovrebbero comprendere sia endpoint di sicurezza (spesso misurati anche in termini di malfunzionamenti tecnici) che endpoint di efficacia clinica. È altrettanto rilevante notare che il beneficio clinico può essere espresso sia relativamente alla salute (o qualità di vita) del singolo paziente che ad un impatto positivo sulla gestione del percorso terapeutico tramite indicatori di Health Technology Assessment (HTA).
In questa prospettiva, vale la pena ricordare la definizione di “beneficio clinico” presente all’Articolo 2.53 del Regolamento MDR:
“l’impatto positivo di un dispositivo sulla salute di una persona, espresso in termini di un esito clinico significativo, misurabile e rilevante per il paziente, ivi compreso l’esito connesso con la diagnosi, ovvero un impatto positivo sulla gestione del paziente o sulla salute pubblica”.
Inoltre, la Meddev 2.7/1 rev 4, pur essendo un documento parzialmente superato da MDCG 2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software, fornisce dettagli tuttora utili per l’identificazione e la valutazione dei “benefici per il paziente” (paragrafo A.72.b), come di seguito riportato:
Evaluation of the device’s benefits to the patient
Positive impacts of a device on the health of an individual should be meaningful (relevant for the patient) and measurable. The nature, extent, probability and duration of benefits should be considered. Benefits may include:
• positive impact on clinical outcome (such as reduced probability of adverse outcomes, e.g. mortality, morbidity; or improvement of impaired body function),
• the patient’s quality of life (significant improvements, including by simplifying care or improving the clinical management of patients, improving body functions, providing relief from symptoms),
• outcomes related to diagnosis (such as allowing a correct diagnosis to be made, provide earlier diagnosis of diseases or specifics of diseases, or identify patients more likely to respond to a given therapy),
• positive impact from diagnostic devices on clinical outcomes, or
• public health impact (such as to the ability of a diagnostic medical device to identify a specific disease and therefore prevent its spread, to identify phases, stages, location, severity or variants of disease, predict future disease onset).
Inoltre, è di grande interesse citare la più aggiornata MDCG 2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software per quanto riguarda alcuni aspetti relativi al beneficio non direttamente misurabile su outcome clinici specifici del paziente.
“Specifically, for MDSW not claiming CLINICAL BENEFITS that can be specified through measurable, patient-relevant clinical outcome(s), clinically relevant outputs are achieved through demonstrated predictable and reliable use and USABILITY (please refer to section 4.2 of this document)”.
• Qualità
Per garantire un livello costante di qualità nel tempo, gli sviluppatori di DTx hanno a disposizione lo standard ISO 13485, Dispositivi medici – Sistemi di gestione della qualità – Requisiti a fini normativi, che è uno standard armonizzato e riconosciuto a livello internazionale, e che stabilisce i requisiti per un sistema di gestione della qualità specifico per l’industria dei dispositivi medici. Una certificazione ISO 13485:2016 non è obbligatoria, ma l’applicazione di questo standard non solo può aiutare le organizzazioni coinvolte in qualsiasi parte del ciclo di vita di un dispositivo medico a dimostrarne la conformità, ma fornisce anche strategie adeguate per la comunicazione a tutti i portatori di interesse.
In particolare, per le DTx, appare centrale la garanzia di poter gestire il ciclo di vita del software, le sue modifiche a fini di correzione (bugfix, manutenzione perfettiva) e/o ai fini di miglioramento incrementale e di poter notificare all’utente qualsiasi modifica, e l’impatto della modifica, sul software.
Risulta adeguato fare riferimento, per questi aspetti tecnici appena citati, nuovamente alla norma IEC 62304, secondo cui (versione bozza) il fabbricante di un dispositivo medico software deve:
“assicurarsi che il software sanitario rilasciato possa essere consegnato in modo affidabile al punto di utilizzo senza corruzione o modifica non autorizzata”.
Inoltre, sempre la IEC 62304 richiede che il fabbricante adotti una procedura per il controllo delle versioni. Nel caso del dispositivo DTx, la politica di versionamento dovrebbe basarsi sull’impatto che la modifica pone sui requisiti normativi. Ad esempio, l’intero sistema DTx potrebbe essere composto da diversi moduli, con versioni SW separate per
• l’algoritmo
• l’interfaccia utente professionale; con versioni diverse in caso di ambienti operativi diversi (ad esempio, l’accesso tramite App Android da un dispositivo intelligente)
• l’interfaccia paziente; con versioni diverse in caso di ambienti operativi diversi (ad esempio, l’accesso tramite App Android da un dispositivo intelligente).
Proponiamo che il controllo delle versioni SW del software rilasciato sia strutturato, coerentemente con la pratica standard per la progettazione software, come SW major.minor.build:
• il numero “major” viene aumentato quando si verificano salti significativi della funzionalità come la modifica del framework che potrebbe causare alterazioni del profilo rischio-beneficio
• il numero “minor” viene incrementato quando sono state aggiunte, corrette o migliorate solo le funzioni minori
• il numero di “build” viene incrementato quando i bug vengono corretti.
Inoltre, il controllo delle versioni può includere la possibilità di avere una versione “release candidate”; in caso di modifica maggiore o minore della versione, la “versione candidata” è la versione SW disponibile solo per i partner selezionati per la valutazione pilota e/o è disponibile solo in un ambiente controllato, separato dall’ambiente commerciale.
La Food and Drug Administration (FDA) propone un approccio basato sul rischio alla modifica del software, indicando la necessità di destinare maggiori risorse per le fasi di verifica e convalida per:
• una modifica che introduce un nuovo rischio o modifica un rischio esistente che potrebbe provocare danni significativi;
• una modifica ai controlli del rischio per prevenire danni significativi;
• una modifica che influisce in modo significativo sulla funzionalità clinica o sulle specifiche delle prestazioni del dispositivo.
Laddove applicato a DTx con moduli di intelligenza artificiale, l’approccio di cui sopra richiederebbe una attenzione speciale quando l’evoluzione del modello:
• influisce in modo significativo sulle prestazioni del dispositivo o sulla sicurezza e l’efficacia
• la modifica impatta per l’uso previsto del dispositivo, ad esempio aumentando la popolazione target
• la modifica introduce importanti modifiche tecnologiche che influenzano le caratteristiche prestazionali.
Inoltre, FDA incoraggia a valutare l’impatto del cambiamento su tre caratteristiche del SaMD:
• prestazioni (cliniche e analitiche)
• input utilizzati dall’algoritmo e loro associazione all’output clinico
• uso previsto, descritto attraverso l’impatto della DTx, sulla condizione di malattia.
1.7. La nostra proposta per un approccio al percorso regolatorio: come sviluppare un software adeguato alla definizione DTx
Il seguente schema propone un approccio alla progettazione del software medicale, adeguato a rispondere ai requisiti regolatori fin dal principio delle attività di progettazione. Si propone quindi di impostare un sistema qualità basato sulla ISO 13485 per garantire una corretta gestione della progettazione del dispositivo medico, integrando una corretta gestione del rischio. Successivamente, si suggerisce di impostare un sistema di gestione del ciclo vita del software secondo la IEC 62304, garantendo quindi una corretta convalida tecnica del software e una corretta gestione dei problemi di sviluppo. Inoltre si propone di integrare nella applicazione della norma IEC 62304 alla progettazione del software anche i principi dettati dalla normativa sulla protezione dei dati personali (GDPR) in tutti i casi in cui questo regolamento sia applicabile. Infine si suggerisce, al termine della fase di sviluppo e di convalida tecnica della sicurezza del dispositivo, di testare l’efficacia tecnica del dispositivo con pazienti simulati.
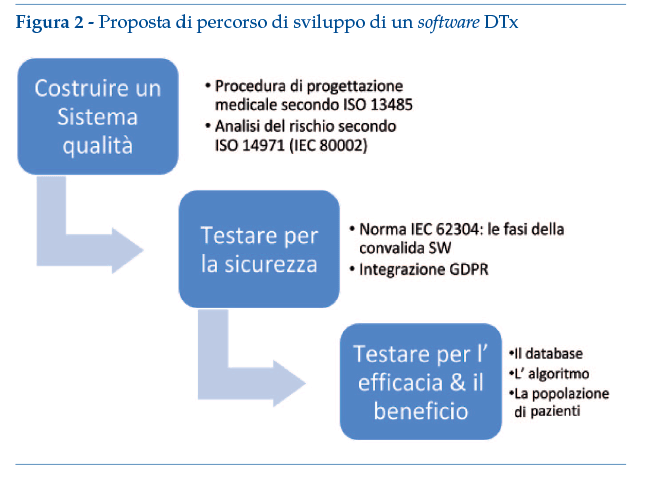
Pertanto, si suggerisce di impostare la progettazione del dispositivo medico software a partire dalla definizione degli input di progetto, come richiesto dalla ISO 13485 e descritto nell’articolo riguardante la “Validazione Tecnica” inserito nel presente volume. Il risultato della fase di progettazione descritta finora dovrebbe essere un elenco di funzionalità del software per ogni profilo utente. I requisiti di progetto così identificati dovrebbero includere i metodi utilizzati per mitigare il rischio per il paziente. Inoltre si suggerisce di coinvolgere i pazienti stessi, a partire dalla fase di definizione degli input, tramite tecniche di progettazione partecipata sia per definire gli user needs che per specificare gli aspetti legati alla esperienza di utilizzo.
Nella definizione delle funzionalità, sarà necessario identificare chiaramente il beneficio clinico atteso: per poter pianificare correttamente gli studi clinici in termini di endpoint misurabili, sarà poi importante definire anche le metriche adeguate a misurare tale beneficio, in coerenza con MDCG 2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software.
Dopo aver definito completamente i requisiti di progetto, sarà necessario passare alla fase di sviluppo e poi alla verifica tecnica. Per questa fase, si propone di testare le funzionalità del dispositivo secondo un piano definito sulla base della lista dei requisiti di input, in modo da poter includere anche la verifica delle misure di minimizzazione del rischio.
Superati i test di convalida delle funzionalità software, in particolare quelli relativi alla sicurezza paziente, si suggerisce di effettuare con un approccio iterativo una valutazione dell’usabilità del dispositivo secondo la IEC 62366, per verificare in ambiente simulato, ma con la partecipazione di utenti reali, che l’interfaccia presenti un adeguato profilo di usabilità.
Tali test, ove il dispositivo debba essere usato anche direttamente dal paziente, dovrebbero coinvolgere anche i pazienti stessi.
Solo successivamente alle verifiche tecniche e di usabilità in ambiente simulato si ritiene adeguato passare alla valutazione dell’efficacia del dispositivo. Prima di passare alla sperimentazione clinica, si suggerisce di completare una validazione tecnica con una fase di simulazione del reale utilizzo clinico, attraverso pazienti definiti appositamente per questo e rappresentativi della popolazione prevista per il dispositivo (Ravizza A.et al. Method for preclinical validation of software as a medical device 2020. DOI: 10.5220/0009155406480655).
Per creare i profili di questi “pazienti simulati” si suggerisce di sfruttare dati disponibili e previo consenso, o reperibili in letteratura. Fra questi dati si suggerisce di identificare le caratteristiche che meglio possano descrivere i pazienti in funzione del dispositivo. Alcuni esempi di caratteristiche possono essere:
• il tempo di reazione dopo l’apparizione di uno stimolo visivo per un dispositivo pensato per la riabilitazione visiva;
• le risposte ai questionari di autovalutazione per un dispositivo per la gestione degli effetti collaterali della chemioterapia;
• le calorie introdotte giornalmente per un dispositivo di trattamento del pre-diabete.
Successivamente, si suggerisce di applicare il principio della moda ai dati associati a queste caratteristiche, in modo da poter creare diversi profili-paziente il più possibile rappresentativi di una reale situazione clinica. Si suggerisce il principio della moda, preferendolo a quello della media, perché quest’ultimo, pur realizzando modelli statisticamente significativi, potrebbe portare alla costituzione di un paziente simulato non rappresentativo di una reale condizione clinica o di reali interazioni con il dispositivo. Per esempio, se nel caso di un questionario si richiedesse ai pazienti di indicare il livello del dolore in una scala da 1 a 5, e tre quarti dei pazienti dichiarassero un dolore eguale a 1, e il quarto rimanente dichiarasse il dolore massimo, applicando il principio della media il paziente simulato esprimerebbe un dolore di valore 2, che non è rappresentativo di nessun paziente. Per questo si suggerisce piuttosto di testare il dispositivo con un paziente simulato che fornisca come risposta 1, in maniera da rappresentare correttamente almeno tre quarti della popolazione dei pazienti.
What is known
• La normativa applicabile
• Le norme tecniche applicabili
• La possibilità di mettere in relazione il concetto di tutela della salute con quello di tutela della privacy e con quello della cybersecurity
• Le criticità e le ambiguità dell’attuale normativa.
What is uncertain
• L’approccio degli Enti Notificati all’applicazione delle norme ai casi DTx, in particolare con intelligenza artificiale.
• In particolare, per quanto riguarda la realtà italiana, rimane aperta la discussione in merito al ruolo nel percorso regolatorio delle Autorità sanitarie, in particolare Ministero della Salute e organi tecnici correlati da un lato, e Agenzia Italiana del Farmaco dall’altro.
What we recommend
• Chiare indicazioni ed esempi per la classificazione in classe I, IIa, IIb e III
• Linee guida sul controllo del versionamento del software, con definizione delle politiche per classificare cambiamenti di versione di tipo major e minor.



