Alice Ravizza1, Enrico Caiani2, Eugenio Santoro3,
Silvia Stefanelli4, Federico Sternini1
1Use-Me-D, Torino
2Politecnico di Milano, Electronics, Information and Biomedical Engineering Department
3Laboratory of Medical Informatics, Mario Negri Institute for Pharmacological Research, Milano
4Stefanelli & Stefanelli Legal Chambers, Bologna-Milano
Tendenze nuove, Numero Speciale 4 2021; 16-36: DOI: 10.32032/TENDENZENUOVENS04202102.PDF

1. Regulatory framework
1.1. Definition of a medical device in Directive 93/42/CEE
A medical device was first technically defined within the European legal system in Directive 93/42/CEE, implemented in Italy by Legislative Decree 46/97.
Article 1 letter a) of Directive 93/42/CEE, in the original version, provided the following definition of a medical device:
“any instrument, apparatus, appliance, material or other article, whether used alone or in combination, including the software necessary for its proper application intended by the manufacturer to be used for human beings for the purpose of:
• diagnosis, prevention, monitoring, treatment or alleviation of disease;
• diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap;
• investigation, replacement or modification of the anatomy or of a physiological process;
• control of conception;
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means.”
In the light of this definition, the software was therefore not considered as conceptually as part and parcel of the medical device in its own right, but only insofar as used for the device’s correct functioning.
The original version of 1993 was thus intended to regulate the so-called “embedded” software, used to run electromedical devices and to provide an interface for them, while the so-called “stand-alone” software was not described.
1.2. Extension of the definition of a medical device in Directive 2007/47/CEE to include software
In 2007 a new Directive (2007/47/CEE – MDD) was issued, providing a more comprehensive definition of a medical device and establishing that software can be a medical device in its own right.
The implications of this modification clearly emerge in recital 6 of the Directive, which states:
“It is necessary to clarify that software in its own right, when specifically intended by the manufacturer to be used for one or more of the medical purposes set out in the definition of a medical device, is a medical device. Software for general purposes when used in a healthcare setting is not a medical device.”
Recital 20 then states:
“Taking account of the growing importance of software in the field of medical devices, be it as stand alone or as software incorporated in a device, validation of software in accordance with the state of the art should be an essential requirement.”
The definition of a medical device is therefore reformulated as follows:
“any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, together with any accessories, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
• diagnosis, prevention, monitoring, treatment or alleviation of disease;
• diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap;
• investigation, replacement or modification of the anatomy or of a physiological process;
• control of conception;
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means.”
Starting from 2007, legislation has thus established that software can be considered a medical device itself, whether it is incorporated in the device (embedded) or works in its own right (stand-alone).
Subsequently, the international IMDRF/SaMD WG/N10FINAL:2013 guidelines introduced the following definition of software as a stand-alone medical device (also called Software as a Medical Device – SaMD):
“The term “Software as a Medical Device” (SaMD) is defined as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.”
1.3. Regulation (EU) 2017/745 on medical devices
In 2017 the new Regulation (EU) 2017/745 on medical devices was approved. Known as the MDR (Medical Device Regulation), this will also be referred to below simply as the Regulation. When it was published in the official Gazette of the European Union on 5 May 2017, the intention was for it to supersede the current Directive and enter into force on 26 May 2020. However, in April 2020, because of the CoViD-19 pandemic, the European Parliament postponed the entry into force of the new Regulation and extended the period of validity of the current Directive by one year, thus establishing the new date of the Regulation’s formal application as 26 May 2021. It should be noted that the Regulation, unlike the Directive, is a full-fledged legislative act, binding in all its parts, which does not need to be transposed into national legislation and thus obliges the member states to comply with it in its entirety.
The new MDR extends the definition of a medical device, including prediction and prognosis among the purposes taken into account, and thus potentially also including software for risk index calculation.
Regarding application of the new rules to software in particular, it is important to point out that Article 2, point 28 of the MDR defines placing on the market as “the first making available of a device, other than an investigational device, on the Union market ”.
Article 5 then states that: “A device may be placed on the market or put into service only if it complies with this Regulation when supplied and properly installed, maintained and used in accordance with its intended purpose.”
Recital 19, with the aim of better defining when software is a medical device, states: “It is necessary to clarify that software in its own right, when specifically intended by the manufacturer to be used for one or more of the medical purposes set out in the definition of a medical device, qualifies as a medical device, while software for general purposes, even when used in a healthcare setting, or software intended for life-style and well-being purposes is not a medical device. The qualification of software, either as a device or an accessory, is independent of the software’s location or the type of interconnection between the software and a device.”So-called wellness apps, meaning those for simple lifestyle tracking, are thus excluded from the definition of a medical device because they are not for “diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of diseases”, while those for tracking and collecting everyday lifestyle data for purposes of primary or secondary prevention do fall within the medical device category.
Regarding the manufacturer’s responsibilities, defined in Article 10 of the Regulation, two of the most important are:
• the need to adopt a system for risk management throughout the medical device’s life cycle, from design to disposal, with a requirement for new, more detailed assessment of post-certification data;
• the obligation to assess the clinical benefits of the product.
Annex 1 of the Regulation includes a list of essential requirements that must be taken into account by the manufacturer for all devices, irrespective of their risk class. To demonstrate conformity with applicable requirements, the manufacturer must adopt different strategies (analyses, tests or procedures): typically, compliance with international/harmonized standards, where available, is considered the most appropriate strategy.
In the case of digital therapeutics, the most appropriate international standards are ISO 13485 and EN ISO 14971 for general requirements, followed by IEC 62304 and IEC 62366 with specific reference to the product’slife cycle and usability.
1.4. Risk classes and medical device software
To identify the medical device’s risk class, its intended use and the related intrinsic risk must be analysed. Identification of the device’s risk class makes it possible to define which strategies and procedures are needed to demonstrate its conformity with the Directive or Regulation. These procedures are designed with a view to resource allocation by the notified bodies and manufacturers, on the principle that high-risk devices require more rigorous testing, while lower-risk devices can be subjected to less costly verification procedures without jeopardizing the patient’s safety.
Medical devices are divided into four risk classes, with the associated degree of risk numbered in ascending order as shown below:
• Class I: less critical devices, such as most non-active and non-invasive devices.
This class comprises three subclasses:
Class Is: devices delivered in a sterile state
Class Im: measuring devices
Class Ir: reusable devices (a new subclass, introduced by the MDR);
• Class IIa: medium-risk devices, such as some non-active devices (both invasive and non-invasive) and active devices that interact with the body in a non-hazardous manner;
• Class IIb: medium-/high-risk devices, such as some non-active devices (especially if invasive) and active devices that interact with the body in a hazardous manner;
• Class III: high-risk devices, such as many implants, those containing drugs or animal derivatives, and some devices that interact with vital organ functions.
Regarding software, both the Directive and the Regulation specify that the classification rules for active devices must be applied. In addition, the Regulation expressly sets out specific rules for software, thus filling a regulatory gap not addressed by the Directive.
The classification rules provided in both the Directive and the Regulation are summarized below, bearing in mind the intention that the former is now superseded by the latter. This reclassification will have important implications, since it will mean that many software products currently included by the Directive in class I will need to be moved into a higher risk class, when reclassification will be necessary.
The Directive sets out the rules for classification in Annex IX, where software is specifically covered by rules 9, 10, 11 and 12. These rules were extensively described and commented on by MEDDEV 2.1/6 of July 2016 and MEDDEV 2. 4/1 Rev. 9 of June 2010 (from which the figure below is reproduced).
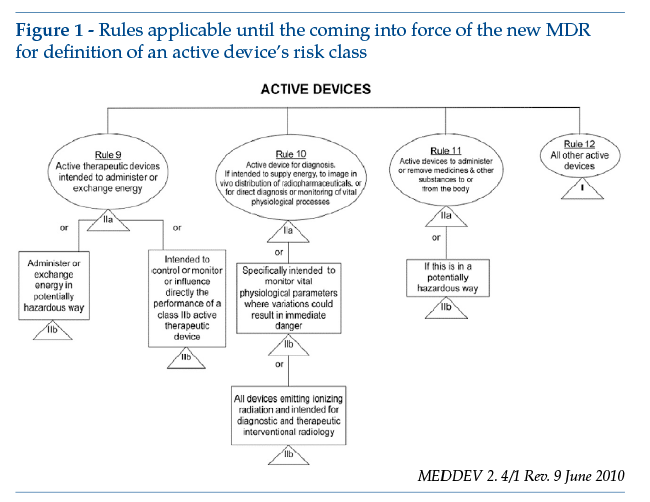
The new classification rules set out in Regulation (EU) 2017/745 can be found in Annex VIII, rule 11:
“Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:
• death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or
• a serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb.
Software intended to monitor physiological processes is classified as classIIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.
All other software is classified as class I.”
During the current period of transition towards application of the new Regulation, there is a clear need to look in greater detail at the different classifications and how they are related to each other, since they all have an impact with a view to handling of post-transition activities. In this regard, the European Commission has recently published a steering document, in the form of specific guidelines on regulatory aspects of software under the MDR and IVDR (MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR): this is seen as a source of guidance in determining whether software falls within the definition of a medical device, as well as in subsequent identification of the corresponding risk class.
The CE marking class, defined on the basis of the certification rules set out in the Regulation, depends on the intended use of the software(when this is consistent with the manufacturer’s recommendations in conditions of normal use). An important implication of this, for the procedure to be followed in obtaining CE marking and placing the software on the market, is the mandatory involvement of a notified body for devices in risk class IIa or higher (meaning, under the new Regulation, practically almost every software fulfilling the definition of a medical device).
The risk class must therefore be determined by analysing the intended use of the software, which can be described in terms of:
• target patient population
• intended users (professional, non-professional, patients)
• clinical performance
• expected clinical benefit.
Briefly stated, risk classes can be broken down as follows for a stand-alone medical device software (Table 1).

1.5. Medical device software classification: some considerations and a proposal
The MDCG 2019/11 ‘Guidance on Qualification and Classification of Software’document, mentioned above, specifies the requirement that the software be used for the benefit of a single individual; this means that it explicitly excludes software used for epidemiological research, or in relation to generic guidelines or clinical protocols that are not patient-specific. On the basis of the MDCG 2019/11 document, it becomes clear that software to be used in managing a single individual’s personal diagnostic procedures and course of treatment is to be considered a “medical device software”, or MDSW. The brief mention of software for planning of treatment pathways, in Decision step 4 (paragraph 3.3), seems to be the only reference in MDCG 2019/11 to the fact that software can perform a therapeutic action. Specifically, the document provides the following comment on rule 11 of the MDR, which is already sufficiently clear and explicit:
“The text of Rule 11 can be divided into what are essentially three subrules that are applied depending on the intended use/purpose of the MDSW:
11a: (3 first paragraphs of Rule 11) intended to provide information which is used to take decisions with diagnostic or therapeutic purposes;
11b: (Paragraph 4 of Rule 11) intended to monitor physiological processes or parameters;
11c: (Paragraph 5 of Rule 11) all other uses.”
Judging by the few specific references in MDGC 2019/11, the authors seem not to have given detailed consideration to the possibility that software may exert a therapeutic action, while the following possible functions are identified:
• data analysis so as to provide a human being, or another software, with information that can be used to take clinical decisions
• monitoring.
MDGC 2019/11 even states the following (paragraph 4.2.1):
“The wording ‘intended to provide information which is used to take decisions with diagnosis or therapeutic purposes’describes, in very general terms, the ‘mode of action ’which is characteristic of all MDSW.”
It thus seems that the legislators are not specifically taking into account the possibility that therapeutic effect is achieved by the software itself, since the feature they identify as common to all MDSW is the function of providing information (processed from input to output).
In addition, it should be noted that not even the many examples provided by the guidelines, with respect to specific types of software and their classification, include any mention of the possibility that they might be used for therapy. The only exception is an example of“Cognitive therapy MDSW that includes a diagnostic function”: this is classified in class III if the diagnostic function is on a closed loop basis, but in class IIa if it provides information to the doctor. Not even in this case, however, is the therapeutic function explicitly considered as a criterion for classification. Another example mentioned is the following: “Diagnostic MDSW intended for scoring depression based on inputted data on a patient’s symptoms (e.g. mood, anxiety) should be classified as class IIb under Rule 11(a)”.
As a result of this, software intended to provide therapy, such as rehabilitative or cognitive-behavioural therapy, could be erroneously placed under the heading “all other uses”and considered as class I.
This interpretation of the classification rules would lead to the risk of classifying all digital therapeutics (DTx) MDSW in class I. We consider that this interpretation is a rapid but dangerous short cut, because it would lead to an erroneous classification in a low risk class for a miscellaneous group of MDSW, including some whose scheduled use has a major impact on the patient’s health.
On the other hand, it should be noted that where a drug in its presentation incorporates a medical device (and, thus, also a SW), whether or not as an integral part, it is defined as a Drug-Device Combination (DDC).
If the drug is an integral partof a device, the relevant part of the MDR is the second sub-paragraph of Articles 1(8) and 1(9):
“1. Devices that when placed on the market or put into service incorporate, as an integral part, a substance that, if used separately, would be considered as a medicinal product, provided that the action of the substance is principal (Article 1(8) MDR). 2. Devices intended to administer a medicinal product, where they form a single integral product intended exclusively for use in the given combination and which is not reusable (Article 1(9) MDR). Typically, these devices have measuring, metering or delivery functions”. The reference document on the subject of drug-device combinations is currently in draft form**.
**29 May 2019 EMA/CHMP/QWP/BWP/259165/2019. Committee for Medicinal Products for Human Use (CHMP). Guideline on the quality requirements for drug-device combinations.
A possible classification of stand-alone DTx can be found in the following table (Table 2) included in MDCG 2019-11, point 11, Annex III. By combining indications of the IMDRF document “Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations”with provisions of Rule 11 – MDR Annex VIII, this table provides operators placing MDSW on the EU market with some useful indicative guidance on the risk class applicable to their products. It is also relevant to point out that this table is actually the only point in the entire document where the word “treat” is explicitly mentioned.
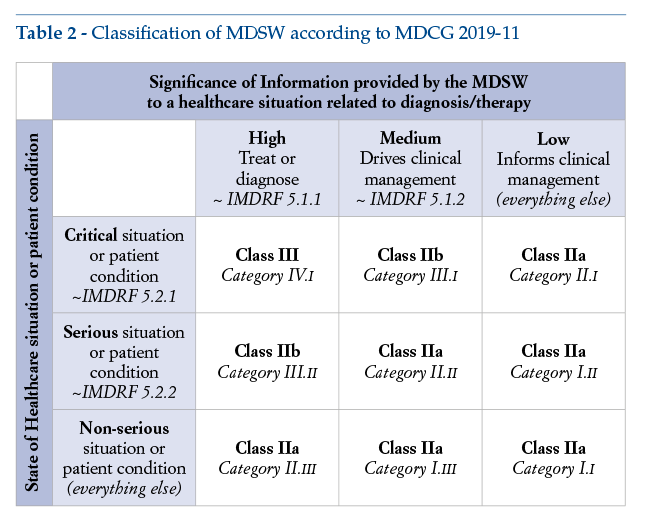
The table shows that therapeutic software is placed in class III, IIb or IIa, according to how critical a condition it is intended to be treated. Class I is not included in the table and, even for the least critical level considered (i.e., for “non-serious”conditions), class IIa is proposed.
This means that, bringing together the different indications stated in the IMDRF framework document’s conversion table and in the EU classification rules as set out in the MDR, the lowest class for any therapeutic software would be class IIa.
The regulatory implications of this are very important, indeed vital. Specifically, only in class I there is provision for the device to receive the CE mark and be placed on the market on the sole basis of a manufacturer’sself-declaration. In all other cases, the Directive and the Regulation specify that, for classes IIa and higher, a notified body must be involved in the certification process.
We therefore find it of great interest that the authors of the Guidelines provide specific examples for DTx, when applied to software with an intended therapeutic function. In addition, it is appropriate that the Guidelines and/or the illustrative examples should provide further comments on the correlation between the severity of the condition to be treated with therapeutic software and the corresponding risk class, incorporating the examples already present in Annex IV.
The classification of DTx into classes IIa, IIb and III, according to the severity of the condition to be treated and the corresponding risk, allows a good degree of consistency between the regulatory requirements already applicable to diagnostic and therapeutic software. Indeed, rule 11 of the MDR establishes medium to high risk classes for monitoring and diagnostic software: the more critical the monitoring or diagnosis, the higher the class.
The model in Table 3 (see below) shows examples of classification for MDSW with a therapeutic function, in order to focus more closely on the peculiar features of DTx.

1.6. Regulatory obligations
Like all medical devices, DTx must respect the essential requirements set out in the current Directive, carried over into the new Regulation but with a number of important updates, above all regarding the obligations in relation to demonstration of clinical benefit.
The manufacturer’s obligations can be summed up in the three keywords “safety”, “benefit” and “quality”. Compliance with these obligations is based on the application of international standards.
•Safety
For demonstration of safety, the reference standard to be consulted by DTx developers is IEC 62304: the version currently in force is dated 2006, but this is at present undergoing widespread and significant revision. It defines methods to design, develop and test software, as well as to manage its entire life cycle, including any updates.
Application of this standard must necessarily go hand in hand with that of ISO 14971, on risk management, dealing in particular with design, testing and updating activities. Application of this standard makes it possible to demonstrate that the software is free of any structural defects constituting a possible risk for the patient, or interfering with its correct functioning in terms of technical performance.
The software’s safety class, defined in compliance with the technical standards set out in IEC 62304, depends on the severity of the damage caused by any breakdown of the software. This class determines the required level of stringency with which the manufacturer must carry out design and functional testing (Table 4).
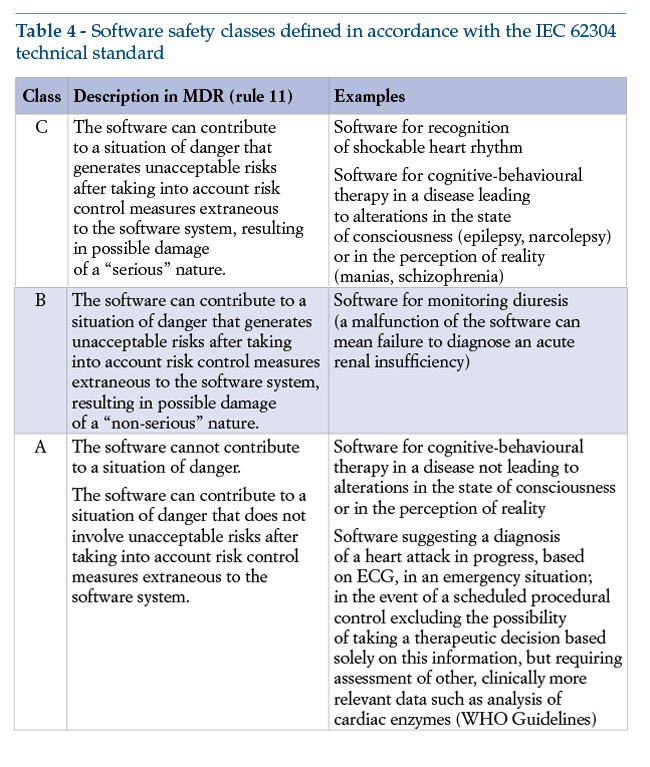
We consider it important to point out that, with a view to the essential criterion of fulfilling requirement 17 of the MDR, the manufacturer must apply management principles to the software’s entire life cycle: these principles are clearly set out in the IEC 62304 standard, the harmonized version of which is EN 62304. Application of harmonized standards makes it possible to presume compliance with essential requirements: this standard thus becomes a technical instrument of major importance for purposes of achieving compliance with the MDR.
It should be emphasized that classification in risk classes from I to III, in accordance with the MDR, and classification in risk classes from A to C, in accordance with IEC 62304, are not formally interrelated. Classification must be established by the manufacturer on a case-by-case basis.
•Benefit
For demonstration of benefit, the reference standard to be consulted by DTx developers is ISO 14155, on good clinical practices for investigation of medical devices. Application of this standard involves demonstrating that, in the target population, the software can achieve clinical benefit significantly outweighing the potential risk involved in its use. In practice, this means moving from indicators of technical performance and safety to indicators of clinical benefit and safety.
Typically, the risk/benefit ratio is investigated in small samples, obviously while ensuring that numbers are appropriate for good statistical evaluation. These studies should include endpoints both for safety (measurement of which often includes any technical malfunction) and for clinical efficacy. It is just as important to note that clinical benefit can be expressed both in terms of health or quality of life in a single patient and also in terms of positive impact on the management of the treatment pathway, as judged by applying Health Technology Assessment (HTA) indicators.
In this perspective, it is worth reiterating the definition of “clinical benefit” in Article 2.53 of the MDR: “the positive impact of a device on the health of an individual, expressed in terms of a meaningful, measurable, patient-relevant clinical outcome(s), including outcome(s) related to diagnosis, or a positive impact on patient management or public health.”
In addition, Meddev 2.7/1 rev 4, though partly superseded by MDCG 2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software, offers guidance that is still relevant with regard to identification and evaluation of “benefits for the patient” (paragraph A.72.b):
Evaluation of the device’s benefits to the patient
Positive impacts of a device on the health of an individual should be meaningful (relevant for the patient) and measurable. The nature, extent, probability and duration of benefits should be considered. Benefits may include:
• positive impact on clinical outcome (such as reduced probability of adverse outcomes, e.g. mortality, morbidity; or improvement of impaired body function);
• the patient’s quality of life (significant improvements, including by simplifying care or improving the clinical management of patients, improving body functions, providing relief from symptoms);
• outcomes related to diagnosis (such as allowing a correct diagnosis to be made, provide earlier diagnosis of diseases or specifics of diseases, or identify patients more likely to respond to a given therapy);
• positive impact from diagnostic devices on clinical outcomes, or
• public health impact (such as to the ability of a diagnostic medical device to identify a specific disease and therefore prevent its spread, to identify phases, stages, location, severity or variants of disease, predict future disease onset).
Also of great interest is the following comment from the more up-to-date MDCG 2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software, covering as it does the question of benefits not measurable through specific clinical outcomes of a patient:
“Specifically, for MDSW not claiming CLINICAL BENEFITS that can be specified through measurable, patient-relevant clinical outcome(s), clinically relevant outputs are achieved through demonstrated predictable and reliable use and USABILITY (please refer to section 4.2 of this document).”
•Quality
To demonstrate consistent quality over a period of time, the reference standard for DTx developers is ISO 13485, Medical devices – Quality management systems – Requirements for regulatory purposes. A harmonized and internationally recognized standard, this document sets out the requirements for a quality management system specifically relevant to the medical devices industry. While an ISO 13485:2016 certification is not mandatory, it not only helps the organizations involved in every stage of a medical device’s life cycle to demonstrate its compliance, but also provides all stakeholders with appropriate communication strategies.
In this respect, a central consideration for DTx is the need to guarantee management of the software’s life cycle, including any corrective modifications(bugfixes, perfective maintenance) and/or incremental improvements, as well as for user notification regarding any modification and its effect on the software.
For the technical considerations identified in the previous paragraph, it is again appropriate to consult the IEC 62304 standard, in the draft version of which the manufacturer of a medical device must:
“ensure that the released health software can be reliably delivered to the point of use, without corruption or unauthorized modification.”
In addition, IEC 62304 also requires that the manufacturer adopt a procedure for control of any subsequent version. For DTx devices, versioning policy should be based on the impact that a version will have on standard requirements. For example, the entire DTx system could comprise various modules, with separate SW versions for:
• the algorithm;
• the professional user interface; with different versions related to different operating environments (for example, access via Android App from a smart device);
• the patient interface; again with different versions related to different operating environments (for example, access via Android apps from a smart device).
We propose structured control over SW versions of the released software, consistent with standard practice for software design, as in major.minor.build semantic versioning:
• the“major”number increases in relation to significant advances in function, like modification of the framework that could alter the risk-
benefit profile;
• the“minor”number increases in relation to additions, corrections or improvements affecting only minor functions;
• the“build”number increases in relation to correction of bugs.
In addition, version control can include the possibility of a “release candidate”version; in the event of the version undergoing a major or minor modification, the “candidate version” is the SW version available only to selected partners for the pilot assessment and/or only in a controlled environment, separate from the commercial environment.
The Food and Drug Administration (FDA) proposes a risk-based approach to modification of software, pointing out the need to ensure that the testing and validation stages benefit from availability of greater resources where a new version involves:
• introduction of a new risk, or modification of an existing risk, with potential to create significant damage;
• modification to risk controls, to prevent significant damage;
• modification significantly affecting the device’s clinical function or performance specifications.
Where applied to DTx with artificial intelligence modules, the above approach would require special attention in cases where any update:
• significantly affects the device’s performance or safety and efficacy;
• impacts the scheduled use of the device, for example by increasing the target population;
• introduces important technical modifications affecting performance characteristics.
In addition, the FDA encourages developers to assess the impact of the change on three characteristics of the SaMD:
• performance (clinical and analytical);
• inputs used by the algorithm, and their association with clinical output;
• scheduled use, as described by the DTx’s impact on the disease condition.
1.7. Our proposal for an approach to regulatory procedures: how to develop software consistent with the definition of DTx
The flow diagram in Figure 2illustrates a suggested approach to medical software design, with a view to ensuring that design activities fulfil regulatory requirements from the very outset. The proposal is therefore to set up a quality system based on ISO 13485, in order to guarantee correct management of medical device designed, including compliance in terms of risk management. After this, it is recommended that a software life-cycle management system be set up according to IEC 62304, thus guaranteeing compliance in terms of the software’s technical validation and correct management of development problems. In addition, when applying IEC 62304 to software design, it is also recommended that the principles of data protection enshrined in GDPR should be fully incorporated where applicable. Finally, when the development phase and the technical validation of the device’s safety have been completed, it is recommended that the device’s technical efficacy be tested with simulated patients.
The first step in setting up medical device software design should therefore be to define the project inputs, as required by ISO 13485 and described elsewhere in this volume (see “Technical validation of digital therapeutics”). The result of the design phase described so far should be a list of the software’s functions for each user profile. Project requirements thus identified should include the methods used to mitigate any risk for the patient.
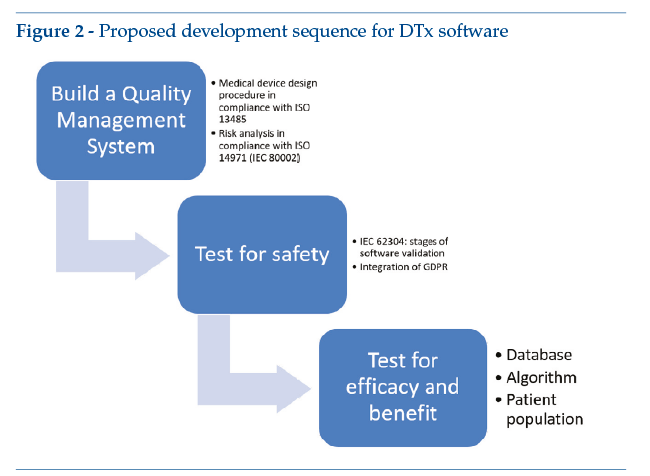
It is also suggested that the patients themselves should be involved, starting from the input definition phase, using participatory project planning techniques both to define user needs and to consolidate user experience.
In defining functions, it will be necessary to clearly identify the expected clinical benefit: for correct planning of clinical trials in terms of measurable endpoints, it will then be important to define appropriate metrics for the measurement of the expected benefits, in compliance with MDCG 2020-1 Guidance on Clinical Evaluation (MDR)/Performance Evaluation (IVDR) of Medical Device Software.
Full definition of project requirements will be followed first by the development phase, and then by technical testing. For this phase, functional testing of the device should follow a plan based on the list of input requirements, thus ensuring that risk minimization measures can also be included.
Once validation testing of software function has been completed, above all in terms of patient safety, the device’s usability should be assessed by an iterative approach in compliance with IEC 62366: this will ensure testing of the interface for an appropriate usability profile, in a simulated environment but with the participation of real users.
Where the device is also intended for direct use by patients, they should be involved in these tests.
Only after technical and usability testing in a simulated environment is it advisable to move on to assessment of the device’s efficacy. Before clinical investigation, it is advisable that technical validation should be completed with simulation of real clinical use, involving patients expressly identified for this purpose and representative of the device’s probable user population (Ravizza A. et al. Method for preclinical validation of software as a medical device 2020. DOI: 10.5220/0009155406480655).
To create the profiles of these “simulated patients”, it is a good idea to use available data (subject, of course, to prior consent) or data from the literature. These data should be used to identify the characteristics that can best describe the patients in relation to the device, including parameters such as:
• reaction time after appearance of a visual stimulus, in the case of a device for visual rehabilitation;
• responses to self-assessment questionnaires, in the case of devices managing side effects of chemotherapy;
• daily calorie intake, in the case of a device for treatment of pre-diabetes.
The statistical measure recommended for these data is the mode, making it possible to create different patient profiles as representative as possible of a real clinical situation. The rationale for preferring the mode to the mean is that the latter, while providing a basis for statistically significant models, could lead to identification of a simulated patient who would not be representative of a real clinical condition or of real interaction with the device. For example, if respondents to a questionnaire are asked to indicate the level of pain on a scale from 1 to 5, and three quarters of them indicate 1 while the remaining quarter indicate 5, the result of applying the statistical mean would be a value of 2 for pain – not actually representative of any single patient. For this reason, it is suggested that the device be tested with a simulated patient giving the answer 1, since this will at least prove representative for three quarters of the patient population.
What is known
• The applicable regulations
• The applicable technical standards
• The possibility of bringing together the concepts of safeguarding health, data protection and cybersecurity
• Critical issues and ambiguities in current regulations.
What is uncertain
• The approach of notified bodies when applying standards to DTx, particularly where artificial intelligence is involved
• In some countries, the role played in regulatory procedures by the health authorities (e.g., in Italy: Ministry of Health and related technical bodies on the one hand, Italian Medicines Agency on the other).
What we recommend
• Clear indications and examples for classification in classes I, IIa, IIb and III
• Guidelines for control of software versions, defining policies for classifying major and minor changes of version.



