Alice Ravizza1, Federico Cabitza2, Enrico Caiani3, Eugenio Santoro4, Federico Sternini1
1Use-Me-D, Torino
2Department of Informatics, Systems and Communication, University of Milano-Bicocca
3Politecnico di Milano, Electronics, Information and Biomedical Engineering Department
4Laboratory of Medical Informatics, Mario Negri Institute for Pharmacological Research, Milano
Tendenze nuove, Numero Speciale 4 2021; 37-50: DOI: 10.32032/TENDENZENUOVENS04202103.PDF

1. Foreword
As is the case with other medical devices, Digital Therapeutics (DTx) too must comply with the essential requirements set out in European standards. These requirements were defined in the current Directive and reconfirmed, albeit with important new features related to the demonstration of clinical benefit, by the new Regulation 2017/745/CE. The requirements as they now stand can be summed up in three keywords: safety,efficacy and quality. Manufacturers can demonstrate that they have fulfilled the obligations set out in the essential requirements by applying international standards.
In this text, the authors focus on the process of development and technical validation of DTx as defined in the ISO 13485 and IEC 62304 standards, up to and including the stage immediately before clinical validation.
2. Fundamentals of technical validation for DTx
Design and technical validation of DTx must be carried out according to the regulatory requirements for medical devices. This means that design must comply with quality system requirements as set out in ISO 13485, and with software life-cycle management requirements as detailed in IEC 62304.
The above-mentioned standards determine the need for consistent and structured definition of clinical needs, and of technical, regulatory and privacy-related constraints, all of which play a decisive role in defining the development planning process for the minimum viable product (MVP) and the final product. The MVP will be used during the various phases of technical validation in order to ensure safety, and will then be brought forward to testing in the field, first for usability and then for the various clinical use pilot phases.
In order for the product to be correctly certified as a medical device, design input requirements must include risk minimization measures consistent with a thorough prior analysis of technical and clinical risk, carried out by the manufacturer in compliance with ISO 14971. A non-exhaustive list of the most useful techniques for DTx risk analysis includes:
• FMEA – failure mode and effects analysis (specifically, inputs defined as per ISO 14971 and IEC/TR 80002-1);
• FTA – fault tree analysis.
These techniques are complementary, with FMEA following an inductive approach while FTA is based on deductive reasoning. On the one hand, FMEA uses initial observation of particulars and identification of failures that have occurred to single components in order to identify system failures; by contrast, FTA begins with general, overall analysis of the type of undesired event (or failure) and uses this as the basis to identify failures of individual components.
Consistent with ISO 13485, development can thus be broken down into a sequence of four main points: definition of intended use, definition of main features, identification of risks, and identification of regulatory constraints. For DTx, each of these four points can be broken down into the subheadings shown below:
- Identification of intended use, in relation to the parameters listed below
a. therapeutic/diagnostic area
b. task/purpose
c. patient population
d. user (patient, caregiver, healthcare professional)
e. claimed benefits, including measurable clinical indicators.
2. Requirements, in terms of function, performance, usability and safety, according to the intended use
a. available functions and their expected results (e.g., delivery of therapy at specific times of day; visualization of state of therapy)
b. technical performance that has to be guaranteed in order to achieve correct use of the device (e.g., how many users at the same time, interaction time)
c. intended forms of user-device interaction (e.g., visualize, select from a list, interact with treatment coordinator, voice command)
d. conditions/situations requiring notification to user (e.g., time almost up)
e. which solutions need to be guaranteed for data protection.
3. Identification of main risks
a. as a result of performance not achieving required levels of accuracy (e.g., failure to achieve appropriate accuracy in measuring a specific parameter, or to recognize a clinically dangerous situation)
b. as a result of software malfunction (e.g., loss of data, or failure to post an “alarm”message)
c. as a result of error in software use (e.g., misinterpretation of an“error” or “alarm”message)
d. other specific risks of the software (e.g., failure to synchronize).
Risks must be identified at this stage, since the software will have to be designed with a view to minimizing both the likelihood of their occurrence and their impact on the patient’s safety. During testing, it will also be necessary to demonstrate that appropriate measures have been taken at the design stage, in order to minimize risk, in the light of regulatory constraints.
4. Identification of major regulatory constraints
a. class of CE mark
b. software risk class as per IEC 62304
c. where applicable: data protection, in compliance with GDPR
d. where applicable: cybersecurity, in compliance with Regulation 2019/881.
3. Minimum required details of DTx to activate the clinical validation phase
3.1. Initial development phase
Initial development techniques for software have not been particularly impacted by regulatory constraints, even if thorough reporting of the various iterations involved can prove helpful for the subsequent validation phase.
DTx manufacturers must nevertheless, even at this stage, determine a number of important indicators for subsequent validation. A non-exhaustive list of these indicators includes the following:
• protective measures against the risk of hacking
• measures for anonymization and pseudonymization of data
• management of the reference hardware (off-the-shelf or dedicated)
• management of any software of unknown provenance (SOUP)
• management of GDPR compliance techniques.
According to IEC 62304, SOUP means software items that are already developed and are generally available, and that have not been developed for the purpose of being incorporated into the medical device (also known as“off-the-shelf”software), or software items previously developed for which adequate records of the development processes are not available.
Some examples of this are the following:
• device drivers
• operating systems (including app operating systems, such as Android and iOS)
• runtimes, including all libraries and programmes that enable running of the software but are not part of the operating system (e.g., virtual machines, libraries for memory allocation).
3.2. Technical verification
The purpose of the technical verification phase is to demonstrate that the software fulfils technical and performance requirements, as specified in the project documentation. The verification phase must be completed in accordance with IEC 62304, the requirements of which become increasingly stringent for higher safety classes.
IEC 62304 subdivides software into three safety classes, according to the severity of damage caused in the event of its failure. Class A comprises software that cannot cause damage; software in class B can cause “non-serious”damage; class C is for software that can lead to “serious”damage.* This means that classification can be carried out by the manufacturer on the basis of case-by-case evaluation and risk analysis. This classification is not related to that found in 2017/745, which is based on the device’s intended use.
*The following definition is proposed:
A serious deterioration in state of health can include (non-exhaustive list from MEDDEV 2.12_1), in accordance with ISO 14155:2020:
a) life-threatening illness,
b) permanent impairment of a body function or permanent damage to a body structure,
c) a condition necessitating medical or surgical intervention to prevent a) or b).
• Examples: clinically relevant increase in the duration of a surgical procedure; a condition that requires hospitalization or significant prolongation of existing hospitalisation.
d) any indirect harm (see definition under section 4.11) as a consequence of an incorrect diagnostic or IVD test result, or as a consequence of the use of an IVF/ART device when used according to the manufacturer’s instructions for use (user errors reportable under section 5.1.5.1 must also be considered).
Tests have to demonstrate the software’s function and performance, in other words its ability to fulfil the requirements established at the stage of needs analysis, including measures to minimize major risks and to fulfil regulatory requirements. Accordingly, technical verification must cover:
• design requirements for individual software elements
• their correct integration
• correct functioning of the software system as a whole
• risk minimization requirements.
3.3. Software architecture and modular certification
Stand-alone software can be subdivided according to its user functions, each of these being related to a module. Functions can be medical or non-medical.
Some examples of non-medical function are the following:
• collection and storage of the patient’s administrative details
• filing of the patient’s medical history.
Some modules can also have ancillary functions, such as:
• audit trail, for reconstruction of events prior to an alarm or audit notification
• access and security, cryptography
• storage and backup of personal data.
The manufacturer must identify the boundaries and interfaces of the different modules, bearing in mind that any module of the software classified as a medical device is subject to certification and regulatory constraints – examples of mandatory MDR requirements being Unique Device Identification (UDI) and post-marketing surveillance.
The limits of modules subject to regulatory requirements for medical devices must be clearly identified by the manufacturer, on the basis of intended use.
If modules subject to regulatory requirements for medical devices are intended for use in combination with other modules within the overall software structure, or with other devices or apparatus, the entire combination must ensure fulfilment of at least the intended function – e.g., a middleware for communication with the hospital system, while not a medical module in its own right, performs its function in combination with the medical module.
These functions must be guaranteed by the software, but are not subject to the life-cycle control requirements set out in IEC 62304: it is nevertheless mandatory to ensure both safety and performance for the overall combination. For example, the module for storage and backup of personal data must guarantee full traceability of the patient.
The manufacturer must evaluate, on the basis of risk analysis, whether segregation measures for the different software modules must be implemented. Segregation is a way of guaranteeing that the software elements do not interact with each other unpredictably, the aim being to avoid side effects from interrelations in the control flow, data flow and access to any shared resources. Segregation works on three different levels:
• Functional: separation of software functions into different modules (e.g., use of middleware);
• Physical: ensuring physical separation of the resources used, by allocation of modules to different hardware units and use of non-shared resources;
• Logical: separation of virtual resources (e.g., use of two different databases).
Segregation makes it possible to establish a separate classification for the different modules that make up the software system, while minimizing propagation of any malfunction together with the associated risk.
The standard makes it possible to use an approach that is at least partly black box in nature, and to carry out tests in a simulated environment. Tests must be performed in ad hocvalidation environments, for all the software’s functional and performance features. For example, it will be possible to create case scenarios representative of typical inpatients in a typical clinic.
The software validation method for regulatory purposes can thus be organized as follows:
- Identify the validation environment as one in which clinical conditions are simulated on the basis of representative cases, appropriate to the type of clinical use scheduled, as in the following examples:
a. creation of an “average patient”, based on data in the literature, characterized by descriptive parameters (clinical anthropometric, phenotypic, behavioural) as identified by a mean-based approach;
b. creation of a “typical”patient, based on data in the literature, characterized by descriptive parameters (clinical, anthropometric, phenotypic, behavioural) as identified by a mode-based approach;
c. creation of a worst case scenario clinical environment – for example, equipped with hardware fulfilling the bare minimum technical requirements.
2. Test software characteristics: in this case, the standard allows a black box approach and requires testing of functional characteristics, risk minimization measures and usability, as well as running a number of typical tests for the device concerned (e.g., installation testing, retrieval from cloud storage). Tests must obviously be repeated for all cases within the purpose-built, simulated environment.
a. Describe the typical flowchart for use of the software. For each functional block (or activity or task), black box testing must be carried out to ascertain that the software can provide a pre-declared and expected deliverable, in terms of the software’s possible interactions and/or output.
b. Integrate ISO 14971 and ISO 80002-1, in order to obtain a list of risks requiring minimization; identify the software characteristics that minimize risk, and subsequently add to the functional tests a number of specific tests to demonstrate correct functioning of minimization measures.
c. Use an approach based on IEC 62366 to test product usability, typically by establishing a list of tasks and focusing on the main ones with user tests.
d. For other specific tests on the product, it can always prove appropriate to describe the functional block beforehand in a flowchart and then test the block, using a black box approach.
3. In the event of failure of one or more tests, investigate the causes with a white box approach, in other words testing implementation of the individual software unit only on the functional block or minimization measure concerned. For example, in the event of an alarm function failing for a vital parameter flagged at a pathological threshold level, investigation should be based on analysis of the software structure: check that the data item correctly completes all the scheduled transformations, and identify the operation or code section responsible for the failure.
3.4. A particular case: machine learning
Machine learning comprises all data-driven techniques for creation of learning- and automatic optimization-based prediction or classification models. By extension and metonymy, the term “machine learning”is also applied to information systems that integrate predictive models created by this technique.
A model’s performance depends on at least two key aspects of design and construction: on the one hand, architecture and training methods; on the other hand, quality and quantity of data. All data used in training must be correctly organized and distributed, so that training can produce a mathematical model adequate for the real data that the software will then be used to analyse.
In the case of models devised and developed to help diagnosis**, data in the training dataset must also be correctly classified (e.g. physiological vs pathological), so that artificial intelligence can be trained to carry out correct classification.
**Consistent with definitions used in the MDR, which includes prognosis and prediction in the definition of medical devices, models of this kind are to all intents and purposes medical devices and therefore require certification based on the appropriate classification.
In order for the algorithm to be as safe, efficient and effective as possible, the training dataset must present the best possible characteristics in terms of quality and reliability. To achieve this objective, data identification must be as clinically correct as is always expected of ground truth data.
There is, however, no universally accepted definition of “clinical truth”, especially in cases where the data item is not produced by a device or a sensor. Far more often, a single, clinically relevant datum regarding an individual patient can be subject to differing interpretations by different doctors, according to their experience or technological literacy: the higher this so-called observer variability, the lower the inter-rater agreement.
Software performance comparable with that of the most skilled human decision-makers may be achieved by training artificial intelligence, with data based on diagnoses and clinical decisions provided by an appropriate number of doctors (at least three, recognized by the scientific community as very experienced or as key opinion leaders – KOL), and with a proven track record for correctly addressing patients’needs and in terms of patient satisfaction.
Correct identification of KOL in terms of skill, knowledge, experience and other characteristics makes it possible to enhance the reliability of the overall result. In some cases, geographical distribution of the experts concerned must also be factored in: European guidelines can be different from US guidelines, while some ethnic groups can be more prone to certain diseases than others, and so on.
For validation of machine learning-based models, the following approaches are useful:
• assess “clinical truth” by comparing the opinions of different KOL on the same data item, giving“truth”status only to a view on which there is complete agreement, or statistically significant consensus (e.g., based on statistical tests like chi-square distribution), or sufficient agreement (in other words, above a certain statistical threshold like Krippendorff’s alpha coefficient);
• create a validation dataset representative of the training dataset in terms of variability and distribution of data regarding the target patient population, or make it as different as possible from the training dataset in order to allow performance assessments in variability-related worst case conditions. Representativeness can be assessed in terms of similarity between datasets by means of statistical tests, such as the multivariate Kolmogorov Smirnov test; in this case, a validation dataset suitable for a worst-case scenario will be associated with a pvalue lower than a given threshold – either 5% or, even more conservatively, 1%. Validation performance below a minimum acceptable accuracy threshold (to be defined on a case-by-case basis) may suggest the need for retraining of the model on more up-to-date or complete data, with a view to their use in continuous monitoring of quality and so-called techno-surveillance of on-market machine learning models;
• maintain a rigid separation of validation and training datasets – in other words, ensure that there are no data in common between the two, and that no transformations (e.g., normalization, standardizations) are made to both on the basis of statistical parameters for only one of the two. This separation of the two datasets, a means of avoiding so-called data leakage, can be achieved by cross-validation techniques such as leave-one-out approaches, provided that precautions are taken to separate the two for every training and performance testing iteration.
The last point is necessary in order to guard against the risk of software malfunction being masked by self-referential validation or, in other words, of the software’s classification performance being invalidated by recognition of data that have already been learnt (a phenomenon known as overfitting).
Among suggested criteria to be fulfilled for compliance with the above indications are the following:
• build validation and training datasets with the same inclusion and exclusion criteria;
• build validation and training datasets representative of the same range of values for parameters associated with the patients included in the dataset (e.g., gender and age distribution, if these are relevant factors for the setting concerned);
• build validation and training datasets representative, where appropriate, of variability in the real patient population, but balanced as far as possible in terms of the classes considered.
Definition of datasets according to these requirements would make it possible to identify the dataset population as the target population for the clinical benefit expected from the medical device, and confirm the suitability of the software for the entire target population. To the same end, the software’s accuracy should also be evaluated by nested cross-validation, making it possible to report accuracy data in terms of proportion of errors and corresponding confidence intervals. The rationale here is that these provide an idea of how far the result obtained on a sample of clinical cases from the reference population can be generalized to other samples from the same population.
Compliance with the above indications makes it possible, at least for functional tests, to treat the software as a black box given the task of classifying a dataset, the clinical truth of which has previously been ascertained by traditional methods. Functional validation can be considered successful if running a test set on the software achieves or surpasses minimum acceptable accuracy, a reference level that can be determined on the basis of the performance required from the device. In particular, determination of minimum acceptable accuracy will also have to depend on assessment of the impact any errors will have on the patient’s health. Finally, a valid basis for its determination can be state-of-the-art specialist literature, based solely on sources covering comparable patient populations. An important consideration in this respect is that performance values will be closely dependent on the data used for validation, which could be identical only if publicly available. After validation, it will be possible to analyse item by item any discrepancies between the doctor’s preliminary evaluation and that given by the software. This analysis can then be used for a further training iteration, in an approach known as active learning.
It should be noted that, in the case of machine learning-based models, clinical validation in a simulated environment will involve considerable difficulties. Often, data based on the literature are either not present or not identifiable; possibly they are available in formats that are difficult to process, unrepresentative of the population or skewed by different biases (e.g., selection, gender, age, ethnic group). In addition, validation on an average case basis would give no guarantee of the technology’s usability with a real population. In this respect, the worst case is not necessarily a technological limitation related to acquisition of the data for analysis, but may be that of an individual differing considerably from the average patient.
Appendix
A.1 Illustrative definition of software architecture and modular certification
The following is an illustrative example of an IT system diagram comprising three main levels (Figure 1):
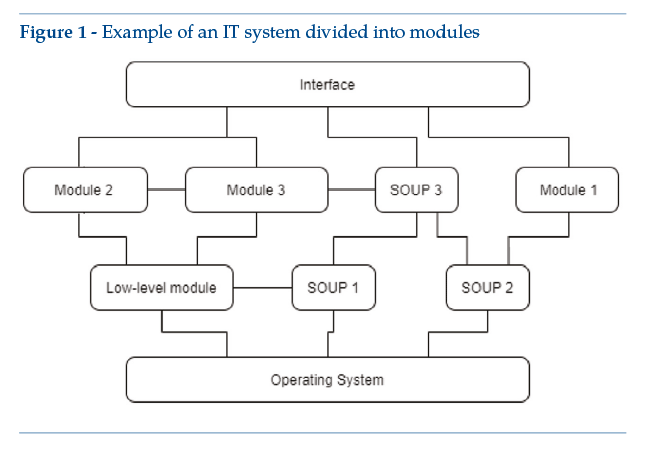
• a level dedicated to interface management between the operating system and the rest of the software;
• an intermediate layer, organized in modules with module-specific software, at least one module being for medical use;
• a top layer, containing the graphic interface and other elements used for module-to-module “connections”.
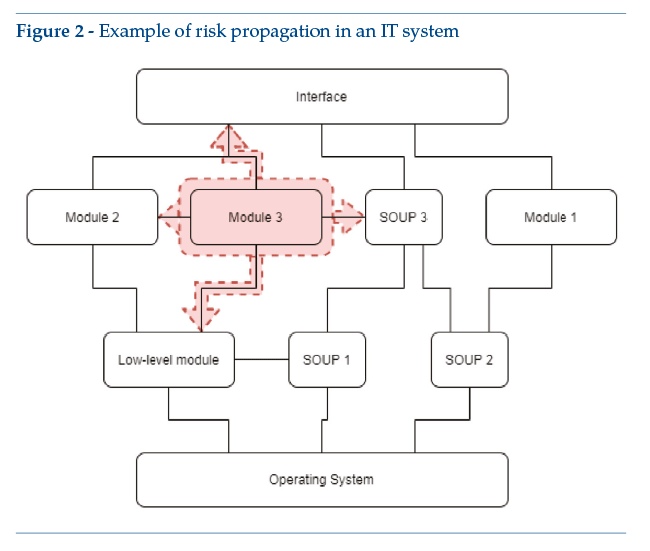
As a concrete example, the above diagram could include an Android app, with enabling SOUP for detection of position, the screen access PIN code and connection to a smartwatch. The app includes self-tracking wellness modules, like a pedometer (module 1) and a sleep tracking function (module 2). Module 3 is functionally different, qualifying as a medical device since it detects epileptic attacks. For the“medical”module, functional limits must be clearly identified – e.g., (by segregation) all functional performance features listed in the system’s scheduled use are managed by module 3. In any case, design constraints for module 3 will have repercussions on other modules or other levels: a case in point is risk analysis, with possible implications for other modules, for the basic levels and for the main graphic interface (Figure 2). If the system contains SOUP affecting or enabling the medical module, medical device design constraints apply.
Conclusions
DTx design must comply fully with the essential requirements typical of medical devices. Knowledge of regulatory requirements for this category of devices makes it possible to have a prior understanding of the importance of software modularity, a fundamental consideration in terms not only of risk and change management but also of critical issues with software of unknown provenance (SOUP) and approaches to their management.
By contrast, there is no consensus to date on how to deal with various aspects of DTx development, such as incremental learning in artificial intelligence and verification in a simulated clinical environment. Incremental learning can be seen both as an opportunity for continuous enhancement of performance and a threat of divergent performance outcomes; while the importance of correctly addressing verification in a simulated clinical environment is recognized in terms of its safety implications, but has yet to be codified in a universally accepted manner.
In this setting, the authors propose integrated test planning to include simulated clinical verification, usability evaluation and verification of risk minimization measures, with a combination of white box and explainable artificial intelligence approaches in the technical validation of the various software elements. This makes it possible to guarantee that patients involved in the clinical validation phase will be exposed to the safest device possible, while also allowing effective risk monitoring.
What is known
• The concept of modular architecture, and its importance in terms of risk and change management
• Management of software of unknown provenance (SOUP).
What is uncertain
• Management of incremental learning in artificial intelligence
• Management of verification in a simulated clinical environment, to determine all safety aspects before the clinical validation phase.
What we recommend
• Integrated test planning, inclusive of simulated clinical verification, usability and verification of risk minimization measures, so as to determine the safety level before the clinical study phase
• As far as possible, use a white box approach
• In creation of artificial intelligence-based devices, use an explainable AI approach, allowing timely identification and mitigation of any risks associated with machine learning algorithms.



