Alice Ravizza1, Federico Cabitza2, Enrico Caiani3, Eugenio Santoro4, Federico Sternini1
1Use-Me-D,Torino
2Dipartimento di Informatica, Sistemistica e Comunicazione, Università degli Studi di Milano-Bicocca
3Dipartimento di Elettronica, Informazione e Bioingegneria, Politecnico di Milano
4Laboratorio di Informatica Medica, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, Milano
Tendenze nuove, Numero Speciale n.01 2021; 37-50: DOI: 10.32032/TENDENZENS20210104.PDF
 Scarica Pdf
Scarica Pdf
1. Premessa
Analogamente a quanto accade per gli altri dispositivi medici, le Terapie Digitali (Digital Therapeutics – DTx) devono rispettare i Requisiti Essenziali previsti dalle norme europee. Questi requisiti sono stati definiti nella attuale Direttiva e riconfermati, pur se con delle importanti novità per quanto riguarda la dimostrazione di beneficio clinico, dal nuovo Regolamento 2017/745/CE. Questi possono essere riassunti da tre parole chiave: sicurezza, efficacia, qualità. I fabbricanti possono dimostrare di aver adempiuto agli obblighi definiti dai Requisiti Essenziali per mezzo dell’applicazione di standard internazionali.
In questo testo gli autori si sono concentrati sul processo di sviluppo e validazione tecnica delle DTx come definito dagli standard ISO 13485 e IEC 62304, fino alla fase immediatamente precedente la validazione clinica.
2. Basi di validazione tecnica della DTx
La progettazione e validazione tecnica di DTx deve essere costruita secondo i requisiti normativi relativi ai dispositivi medici. Pertanto la progettazione deve rispettare i dettami della norma ISO 13485 relativa ai sistemi di qualità, e della norma IEC 62304 relativa alla gestione del ciclo di vita del software.
Le suddette norme determinano la necessità di definire in modo coerente e strutturato i bisogni clinici, i vincoli tecnici, regolatori e di privacy, determinanti per definire il processo di progettazione dello sviluppo del Minimum Viable Product (MVP) e del prodotto finale. Il MVP sarà utilizzato nelle fasi di validazione tecnica per assicurarne la sicurezza e sarà poi avviato a una fase di test sul campo, prima per la usabilità e poi per le fasi pilota di uso clinico.
Perché il prodotto possa essere correttamente certificato come dispositivo medico, i requisiti di input della progettazione devono includere le misure di minimizzazione del rischio identificate per mezzo di una corretta analisi del rischio tecnico e clinico, effettuata dal fabbricante seguendo la norma ISO 14971. Le tecniche più utili per la analisi di rischio di una DTx includono, ma non sono limitate a:
• FMEA – “analisi dei modi e degli effetti dei guasti“, dall’inglese Failure Mode and Effect Analysis (in particolare, input di ingresso definiti in coerenza con la norma ISO 14971 e la norma IEC/TR 80002-1);
• FTA – “albero dei guasti”, dall’inglese Fault Tree Analysis.
Queste tecniche sono complementari, la prima induttiva e la seconda deduttiva, in quanto la FMEA parte dal particolare e dalla identificazione di guasti occorsi a livello dei singoli componenti per giungere all’identificazione di guasti a livello di sistema, mentre la FTA parte da un’analisi generale e complessiva del tipo di eventi indesiderati (o guasti) e arriva ad identificare i guasti a livello dei singoli componenti.
Lo sviluppo dunque può essere strutturato, secondo la norma ISO 13485, per quattro punti principali: la definizione dell’uso previsto, la definizione delle principali caratteristiche, l’identificazione dei rischi e l’identificazione dei vincoli regolatori. Si propone, per ciascun punto, un approfondimento specifico per il settore DTx, come segue:
1. Identificazione dell’uso previsto
a. quale area terapeutica/diagnostica
b. quale compito/finalità
c. quale popolazione di pazienti/assistiti
d. quale utilizzatore (paziente, caregiver, professionista sanitario)
e. quale claim di beneficio, comprensivo degli indicatori clinici misurabili.
2. Requisiti funzionali, prestazionali, di usabilità e di sicurezza, in base all’uso previsto
a. funzionalità disponibili e corrispondenti risultati attesi (ad esempio: funzionalità di erogazione della terapia in momenti specifici della giornata; visualizzazione dello stato della terapia)
b. quali prestazioni tecniche devono essere garantite per realizzare il corretto utilizzo del dispositivo (ad esempio: quanti utenti in contemporanea, tempo di interazione)
c. quali sono le interazioni previste dell’utente con il dispositivo (ad esempio visualizza, seleziona da una lista, interagisce con gestore, comando vocale)
d. quali condizioni devono essere notificate all’utente (ad esempio: il tempo sta per scadere)
e. quali soluzioni devono essere garantite per la protezione dei dati.
3. Identificazione dei principali rischi
a. in conseguenza di una prestazione insufficiente (ad esempio, se la accuratezza nella misura di un parametro non è adeguata oppure se una situazione clinicamente pericolosa non viene riconosciuta)
b. in conseguenza di un malfunzionamento del software (ad esempio, perdita dati o mancato allarme)
c. in conseguenza di un errore di utilizzo del software (ad esempio, una errata interpretazione di un messaggio di errore o di allarme)
d. altri rischi specifici del software (ad esempio, mancata sincronizzazione).
I rischi devono essere identificati in questa fase, poiché sarà necessario progettare il software per poterne minimizzare la probabilità di occorrenza e l’impatto sulla sicurezza del paziente. Inoltre sarà necessario, in fase di verifica, dimostrare come nella progettazione siano state prese adeguate misure di minimizzazione dei rischi, alla luce dei vincoli regolatori.
4. Identificazione dei principali vincoli regolatori
a. classe di marcatura CE
b. classe di rischio software secondo IEC 62304
c. eventualmente: privacy secondo il GDPR
d. eventualmente: cybersecurity secondo il Regolamento 2019/881.
3. Minimo dettaglio necessario della DTx per attivare
la fase di validazione clinica
3.1. La fase di sviluppo iniziale
Le tecniche di sviluppo iniziale del software non sono particolarmente impattate dai vincoli regolatori, anche se una corretta rendicontazione delle varie iterazioni può aiutare la successiva fase di convalida.
I produttori di DTx devono però, già in questa fase, determinare alcuni importanti indicatori per la convalida successiva. Questi indicatori includono, ma non sono limitati a:
• misure di protezione dal rischio di hackeraggio
• misure di anonimizzazione o pseudonimizzazione del dato
• gestione dell’hardware di riferimento (commerciale o dedicato)
• gestione di eventuali software di origine esterna, i cosiddetti SOUP (Software of Unknown Provenance)
• gestione delle tecniche di compliance con la GDPR.
Si definiscono SOUP, secondo la IEC 62304, gli elementi software precedentemente sviluppati, e non sviluppati per essere integrati nel dispositivo medico.
Si citano come esempi:
• i driver degli hardware associati
• i sistemi operativi (che comprendono anche i sistemi operativi delle app, quali Android e iOs)
• i runtimes, che comprendono tutte le librerie e i programmi che forniscono servizi per l’utilizzo del software, senza essere parte del sistema operativo (per esempio le macchine virtuali o le librerie per l’allocazione della memoria).
3.2. La fase di verifica tecnica
La fase di verifica tecnica (verification) è atta a dimostrare che il software soddisfa i requisiti tecnici e di prestazione (performance), come questi sono stati specificati nella documentazione di progetto. La fase di verifica deve essere portata a termine secondo le indicazioni della IEC 62304, i cui requisiti si fanno più stringenti per i prodotti delle classi superiori di sicurezza.
La norma tecnica IEC 62304 suddivide il software in tre classi di sicurezza sulla base della gravità del danno causato nel caso di un guasto del software stesso. Alla classe A sono associati software che non possono causare danni, alla classe B software che possono causare danni “non seri”, e alla classe C software che possono portare a danni “seri”1.
1 Si propone la definizione
A serious deterioration in state of health can include (non exhaustive list from MEDDEV 2.12_1) and compliant to ISO 14155:2020:
a) life-threatening illness,
b) permanent impairment of a body function or permanent damage to a body structure,
c) a condition necessitating medical or surgical intervention to prevent a) or b).
• Examples: – clinically relevant increase in the duration of a surgical procedure, – a condition that requires hospitalisation or significant prolongation of existing hospitalisation.
d) any indirect harm (see definition under section 4.11) as a consequence of an incorrect diagnostic
or IVD test result or as a consequence of the use of an IVF/ART device when used within Manufacturer’s instructions for use (use errors reportable under section 5.1.5.1 must also be considered).
Pertanto, la classificazione deve essere eseguita dal fabbricante caso per caso e sulla base dell’analisi di rischio. Questa classificazione non è correlata con quella caratteristica del Regolamento 2017/745 che invece è basata sulla destinazione d’uso del dispositivo.
I test devono dimostrare la funzionalità e la performance del software, cioè la capacità di soddisfare i requisiti stabiliti in fase di analisi dei bisogni, comprendendo anche misure per minimizzare i rischi principali e per soddisfare i requisiti regolatori. Pertanto, le verifiche tecniche devono coprire:
• I requisiti di progetto dei singoli elementi software
• La corretta integrazione degli elementi software
• La funzionalità del sistema software nel suo insieme
• I requisiti di minimizzazione del rischio.
3.3. L’architettura del software e la certificazione modulare
I software stand-alone possono essere suddivisi in base alle funzionalità per l’utente, in cui ciascuna di queste funzionalità è correlata a un modulo. Le funzionalità possono essere mediche o non mediche.
Alcuni esempi di funzionalità senza scopo medico sono:
• raccolta e conservazione dei dettagli amministrativi del paziente
• archiviazione della storia medica del paziente.
Alcuni moduli possono inoltre avere funzioni ancillari, si citano ad esempio:
• audit trail, per la funzionalità di ricostruzione degli eventi precedenti a un allarme o a una notifica
• access and security, crittografia
• archiviazione e backup dati anagrafici.
È obbligo del fabbricante identificare i confini e le interfacce dei diversi moduli, tenendo in considerazione che qualora un modulo del software sia classificato come dispositivo medico, tale modulo è soggetto alla certificazione e ai vincoli regolatori, inclusi ad esempio per il MDR la assegnazione di UID (Unique Identification Device) e la post-marketing surveillance.
I limiti dei moduli soggetti ai requisiti regolatori sui dispositivi medici devono essere chiaramente identificati dal produttore e basati sull’uso previsto.
Se i moduli soggetti ai requisiti regolatori sui dispositivi medici sono destinati all’uso in combinazione con altri moduli dell’intera struttura del software, altri dispositivi o apparecchiature, l’intera combinazione deve assicurare almeno la funzionalità prevista. Ad esempio, un middleware di comunicazione con il sistema ospedaliero non è di per sé un modulo medicale, ma svolge la sua funzione in combinazione con il modulo medicale.
Tali funzioni devono essere garantite dal software, ma non sono soggette ai requisiti di controllo del ciclo di vita previsti nella norma IEC 62304: è necessario comunque assicurare sia la sicurezza che le prestazioni, relativamente all’intera combinazione. Ad esempio, il modulo di archiviazione e backup dei dati anagrafici deve garantire la completa tracciabilità del paziente.
Il fabbricante deve valutare, secondo l’analisi di rischio, se implementare misure di segregazione dei moduli software. La segregazione è un modo per garantire che gli elementi software non si influenzino reciprocamente in modo imprevisto, e ha lo scopo di evitare gli effetti collaterali derivanti dalle interrelazioni nel flusso di controllo, nel flusso di dati e nell’accesso ad eventuali risorse condivise. La segregazione funziona su tre diversi livelli:
• Funzionale: separazione delle funzionalità del software in diversi moduli (ad esempio l’utilizzo di middleware)
• Fisica: separazione fisica delle risorse impegnate dai moduli (ad esempio l’allocazione dei moduli su diversi hardware e la non condivisione di risorse)
• Logica: separazione delle risorse virtuali (ad esempio utilizzo di due database diversi).
La segregazione consente di classificare separatamente i diversi moduli che compongono il sistema software e di minimizzare la propagazione di malfunzionamenti e del rischio connesso.
La norma consente di utilizzare un approccio almeno parzialmente “a scatola nera” (black-box) e di eseguire i test in ambiente simulato. I test dovrebbero essere svolti in ambienti di validazione ad hoc per testare tutte le caratteristiche funzionali e prestazionali del software. Ad esempio, sarà possibile creare degli scenari di caso (case scenario), con “pazienti tipo” ricoverati in una “clinica tipo”.
Il metodo per la convalida di software ai fini regolatori può quindi essere organizzato come segue:
1. Identificare l’ambiente di validazione come un ambiente in cui le condizioni cliniche siano simulate tramite casi tipici, adeguati al tipo di uso clinico previsto, per esempio:
a. creazione del “paziente medio” basato su dati di letteratura, caratterizzato appunto da parametri descrittivi (clinici, antropometrici, fenotipici, comportamentali) identificati con il principio della media
b. creazione del “paziente tipo” basato su dati di letteratura, caratterizzato da parametri descrittivi (clinici, antropometrici, fenotipici, comportamentali) identificati con il principio della moda
c. creazione dell’ambiente clinico nel caso peggiore (worst case), per esempio dotato di hardware con i minimi requisiti tecnici richiesti.
2. Testare le caratteristiche del software: la norma consente in questo caso un approccio black-box e richiede di testare le caratteristiche funzionali, le misure di minimizzazione del rischio, la usabilità, ed alcuni altri test tipici del singolo dispositivo (ad esempio test di installazione, di trasferimento dai cloud). Chiaramente i test saranno da ripetere per tutti i casi dell’ambiente simulato, creato precedentemente.
a. Descrivere la flowchart di utilizzo tipico del software e testare, per ciascun blocco funzionale (o attività o task) in modalità “black box” la capacità del software di fornire un deliverable atteso pre-dichiarato, in termini di possibili interazioni e/o output del software
b. Integrare la norma ISO 14971 e la norma ISO 80002-1 per ottenere una lista di rischi che necessitano di minimizzazione; identificare le caratteristiche software che minimizzano il rischio e successivamente aggiungere ai test funzionali alcuni test specifici per dimostrare il funzionamento delle misure di minimizzazione
c. Utilizzare un approccio secondo la norma IEC 62366 per testare l’usabilità del prodotto, tipicamente tramite una lista di task e alcuni user test focalizzati su task principali
d. Per altri test specifici del prodotto, può sempre essere adeguato descrivere dapprima in flowchart il blocco funzionale, e poi testare il blocco con approccio black-box.
3. In caso di fallimento di uno o più test, indagare le cause con approccio white box, che prevede di testare l’implementazione della singola unità software solo sul blocco funzionale oppure sulla misura di minimizzazione in questione. Per esempio, nel caso di fallimento di una funzionalità di allarme relativa ad un parametro vitale, che ne notifichi un valore in soglia patologica, si suggerisce di indagare analizzando la struttura del software: verificare che il dato completi correttamente tutte le trasformazioni previste ed identificare l’operazione o la sezione di codice che ha portato al fallimento.
3.4. Un caso particolare: machine learning
Con machine learning si intendono tutte le tecniche data-driven di realizzazione di modelli di predizione o classificazione basate su apprendimento e ottimizzazione automatica. Per estensione e metonimia, con il termine machine learning ci si riferisce anche ai sistemi informatici che integrano modelli predittivi costruiti con questa tecnica.
Le prestazioni di un modello dipendono da almeno due caratteristiche cardine di progetto e realizzazione: architettura e metodi di addestramento da una parte, qualità e quantità dei dati dall’altra. I dati usati in addestramento devono essere correttamente organizzati e distribuiti in modo che l’addestramento produca un modello matematico adeguato ai dati reali che in seguito il software si troverà ad analizzare.
Nel caso di modelli pensati e sviluppati per l’assistenza alla diagnosi2, i dati del dataset di addestramento (training) devono essere anche correttamente classificati (ad esempio, fisiologico vs patologico), affinché l’intelligenza artificiale si possa addestrare a effettuare la corretta classificazione.
2 In conformità alle definizioni presenti in MDR che inserisce prognosi e predizione alla definizione di dispositivi medici, tali modelli sono a tutti gli effetti dei dispositivi medici, e devono quindi essere certificati in base alla opportuna classificazione.
Perché l’algoritmo sia il più sicuro, efficiente ed efficace possibile, il dataset di training dovrà presentare le migliori caratteristiche possibili in termini di qualità e affidabilità (reliability). Per realizzare questo obiettivo, i dati devono essere identificati in maniera clinicamente corretta, come in una sorta di “verità clinica” (ground truth) di riferimento.
La “verità clinica” tuttavia non ha una definizione univoca, soprattutto in quei casi in cui il dato non è prodotto da un macchinario o da un sensore: molto più spesso un singolo dato di rilevanza clinica che riguarda un singolo paziente può essere valutato diversamente da medici differenti, in funzione di diversi livelli di esperienza o diverse competenze nell’uso della tecnologia: è il fenomeno noto con l’espressione “observer variability”, che è tanto maggiore quanto minore è il cosiddetto “inter-rater agreement”.
Addestrare l’intelligenza artificiale usando diagnosi e decisioni cliniche di un numero adeguato (almeno tre) di medici che siano riconosciuti dalla comunità scientifica come molto esperti o “key opinion leaders” (KOL), che siano fra l’altro anche in grado di valorizzare i bisogni e il gradimento dei pazienti, può consentire di ottenere prestazioni del software confrontabili con quelle dei più esperti esseri umani.
La corretta identificazione dei KOL in termini di competenza, conoscenza, esperienza e altre caratteristiche consente di migliorare l’affidabilità del risultato complessivo. Si pensi ad esempio a come sia in alcuni casi anche necessario considerare la distribuzione geografica di tali esperti: linee guida europee possono essere diverse da quelle americane, certe etnie possono essere più prone a sviluppare certe patologie e non altre, eccetera.
Per la validazione di modelli basati su machine learning si suggerisce:
• stimare la “verità clinica” per mezzo del confronto delle opinioni di diversi KOL rispetto allo stesso dato, mantenendo come “verità” solo quella in cui c’è totale accordo, o consenso statisticamente significativo, ad esempio sulla base di test statistici come quello del Chi Quadro, oppure di un accordo sufficiente, cioè superiore ad una certa soglia in termini di una misura statistica di accordo, ad esempio l’Alfa di Krippendorff;
• la realizzazione di un dataset di validazione rappresentativo del dataset di training in termini di variabilità e distribuzione del dato, per quanto riguarda la popolazione di pazienti target, oppure che sia il più possibile diverso dal dataset di validazione per permettere valutazioni di worst case di variabilità. La rappresentatività può essere valutata in termini di similarità tra dataset per mezzo di test statistici, come ad esempio il test multivariato di Kolmogorov Smirnov; in questo caso, un dataset di validazione adatto ad uno scenario di “caso peggiore” sarà quello associato ad un p-value inferiore ad una determinata soglia, o il 5% o, in maniera ancora più conservativa, l’1%. Prestazioni di validazione inferiori ad una determinata soglia, la cosiddetta minimum acceptable accuracy (da definire caso per caso), potranno suggerire la necessità di riaddestrare il modello su dati più aggiornati o completi e quindi essere usati in un contesto di monitoraggio continuo della qualità e di “tecno-vigilanza”;
• la rigida separazione del dataset di validazione dal dataset di training, cioè il requisito che non vi siano dati in comune fra questi due dataset né che siano fatte trasformazioni su entrambi gli insiemi di dati, quali ad esempio normalizzazione o standardizzazioni sulla base di parametri statistici di uno dei due (per evitare il fenomeno detto di data leakage), è possibile utilizzando tecniche di convalida incrociata, ad esempio approcci leave-one-out, purché le accortezze di separazione fra dataset di training e di validazione siano mantenute per ogni iterazione di allenamento e verifica delle prestazioni.
L’ultimo punto è necessario per evitare il rischio che un malfunzionamento del software sia mascherato da una validazione “autoreferenziale”, o in altre parole, che le performance di classificazione del software siano inficiate dal riconoscimento di dati già appresi, per il noto fenomeno dell’overfitting.
Si suggerisce un esempio di criteri da rispettare per essere conformi alle indicazioni precedentemente elencate:
• costruire dataset di training e validazione con i medesimi criteri di inclusione ed esclusione;
• costruire il dataset di validazione ed il dataset di training tali da essere rappresentativi dello stesso intervallo di valori dei parametri associati ai pazienti inclusi nei dataset; ad esempio, particolare attenzione va posta nella distribuzione per sesso e per età, se per l’ambito di applicazione tali fattori hanno rilevanza;
• costruire il dataset di validazione ed il dataset di training tali da essere, dove adeguato, rappresentativi della variabilità nella popolazione reale di pazienti, ma il più possibile bilanciati nelle classi considerate.
La definizione dei dataset secondo questi requisiti consentirebbe di identificare la popolazione del dataset come la popolazione target del beneficio clinico atteso del dispositivo medico, e confermare l’adeguatezza del software per l’intera popolazione target. Allo stesso scopo, si consiglia di valutare la accuratezza del software anche attraverso nested crossed validation (convalida incrociata annidata) in maniera tale da poter riferire dati di accuratezza in termini di proporzioni di errori e i relativi intervalli di confidenza, in quanto questi ultimi forniscono una idea di quanto il risultato ottenuto su un campione di casi clinici estratti dalla popolazione di riferimento possa essere generalizzato ad altri campioni provenienti dalla medesima popolazione.
Il rispetto delle indicazioni elencate consente, almeno per i test funzionali, di approcciare il software come una scatola nera/black box a cui viene dato il compito di classificare un set di dati, la cui “verità clinica” è stata precedentemente accertata con metodi tradizionali. La validazione funzionale si potrà dire conclusa con successo se l’accuratezza del software (valutata sul test set) sarà almeno pari ad una minimum acceptable accuracy, un livello di riferimento che potrà essere determinato sulla base delle prestazioni desiderate dal dispositivo. In particolare la stima della minimum acceptable accuracy dovrà dipendere anche dalla valutazione dell’impatto di eventuali errori sulla salute del paziente. In ultimo, per la definizione di tale valore sarà possibile, secondo il principio di non inferiorità con lo stato dell’arte, fare ricorso alla letteratura specialistica (stato dell’arte), facendo riferimento esclusivamente a fonti che siano relative a popolazioni di pazienti comparabili. È necessario ricordare che in quest’ultimo caso i valori delle prestazioni saranno strettamente dipendenti dai dati utilizzati per la validazione, che potrebbero essere identici solo nel caso di dati pubblicamente disponibili. Successivamente, si potranno analizzare dato per dato eventuali scostamenti tra la valutazione data preliminarmente dal medico e quella data dal software, ed utilizzare questa analisi per una nuova iterazione di addestramento, nell’approccio noto con l’espressione di active learning.
È da notare che, in caso di modelli basati sul machine learning, l’approccio di validazione clinica in ambiente simulato presenta difficoltà elevate, poiché i dati di letteratura sono spesso non presenti o non reperibili, o sono disponibili in formati difficili da elaborare, non rappresentativi della popolazione, o deformati da diversi bias, tra i quali quello di selezione, genere, anagrafico e razziale. Inoltre, validare sul caso medio non darebbe nessuna garanzia di utilizzabilità della tecnologia su una popolazione reale. Il worst case, in questa logica, non è solo un limite tecnologico nella fase di acquisizione dei dati da analizzare, ma un paziente particolarmente diverso dal paziente medio.
Appendice
A.1 Esempio di definizione di architettura del software
e certificazione modulare
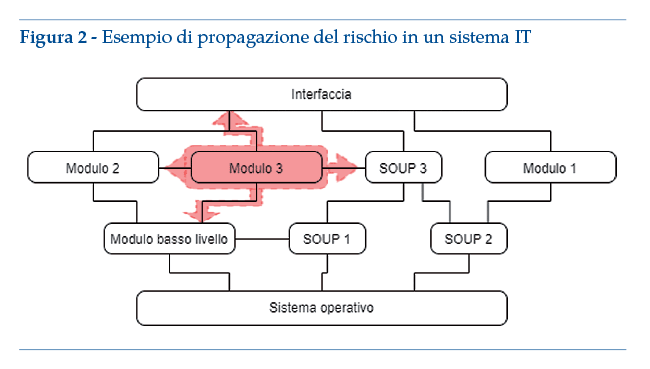
Si presenta a titolo esemplificativo uno schema di sistema IT composto da 3 livelli principali:
• un livello dedicato alla gestione di aspetti di interfacciamento fra resto del software e sistema operativo;
• uno strato intermedio organizzato in moduli, che contengono software specifici per modulo; di questi almeno un modulo ha uso previsto medicale;
• uno strato superiore che contiene l’interfaccia grafica e altri elementi per “collegare” i moduli l’un l’altro.
Come esempio concreto, lo schema sopra riportato potrebbe comprendere una App in Android, con annesse SOUP di riconoscimento della posizione e del PIN di accesso allo schermo. Inoltre sono presenti delle SOUP per collegarsi a uno smartwatch. L’App presenta dei moduli di self-tracking per il benessere, ad esempio il contapassi (modulo 1) e il riconoscimento delle fasi di sonno (modulo 2). Il modulo n.3 rientra invece nella qualifica di dispositivo medico in virtù della sua funzione di riconoscimento di attacchi epilettici del paziente. Per il modulo “medicale” devono essere chiaramente identificati i limiti funzionali, per esempio (tramite segregazione) tutte le prestazioni funzionali elencate nell’uso previsto sono gestite dal modulo 3. In ogni caso, i vincoli di progettazione del modulo 3 avranno ripercussioni su altri moduli o altri livelli. In particolare, l’analisi del rischio può avere conseguenze su altri moduli, sui livelli di base e sull’interfaccia grafica principale. Se il sistema contiene SOUP che influenzano o permettono l’utilizzo del modulo medicale, vanno adottati i vincoli di progettazione dei dispositivi medici.
Conclusioni
La realizzazione di DTx richiede che la progettazione del dispositivo rispetti i requisiti essenziali tipici dei dispositivi medici. Attualmente la conoscenza dei requisiti regolatori per questa categoria di dispositivi consente di avere una conoscenza pregressa sull’importanza della modularità dei software, fondamentale aspetto da valutare in termini di gestione del rischio e del cambiamento, sulle criticità derivanti dall’utilizzo di software di terze parti (Software Of Unknown Provenance – SOUP) e sugli approcci per la gestione di tali criticità.
In contrasto, non c’è ancora un consenso riguardo alcuni temi relativi allo sviluppo di DTx come la gestione dell’apprendimento incrementale delle intelligenze artificiali, che si presenta in contemporanea come promessa di continuo miglioramento delle prestazioni e minaccia di divergenza di quest’ultime, e la gestione delle verifiche in ambiente clinico simulato, di cui si riconosce l’importanza per la determinazione degli aspetti di sicurezza, ma di cui manca ancora una codifica univoca.
In questo contesto gli autori propongono di realizzare dei piani di test integrati che includano la verifica clinica simulata, la valutazione dell’usabilità e la verifica delle misure di minimizzazione del rischio, combinando, quando possibile, approcci white box e di Explanable Artificial Intelligence nella validazione tecnica dei vari elementi del software, in maniera tale da garantire ai pazienti che verranno coinvolti nella fase di validazione clinica il dispositivo più sicuro possibile, e al tempo stesso di consentire di effettuare un efficace monitoraggio dei rischi.
What is known
• Il concetto di architettura modulare e la sua importanza in termini di gestione del rischio e gestione del cambiamento
• La gestione dei SOUP (Software of Unknown Provenance)
What is uncertain
• La gestione dell’apprendimento incrementale delle intelligenze artificiali
• La gestione delle verifiche in ambiente clinico simulato, per determinare tutti gli aspetti di sicurezza prima della fase di validazione clinica
What we recommend
• Piano di test integrato composto da verifica clinica simulata, usabilità e verifica delle misure di minimizzazione del rischio per determinare il livello di sicurezza, prima di accedere agli studi clinici
• Utilizzare per quanto possibile un approccio white box
• Nel contesto della realizzazione di dispositivi basati su intelligenza artificiale, sfruttare un approccio di Explainable Artificial Intelligence, in maniera tale da permettere di identificare tempestivamente e mitigare eventuali rischi introdotti dagli algoritmi di machine learning.



